CHAPTER 58 Acute Pancreatitis
INCIDENCE AND BURDEN OF DISEASE
The incidence of acute pancreatitis in England, Denmark, and the United States varies from 4.8 to 38 per 100,000 patients.1–3 However, estimates of incidence are inaccurate because the diagnosis of mild disease may be missed, and death may occur before diagnosis in 10% of patients with severe disease.4
Diseases of the pancreas (acute and chronic pancreatitis) accounted for 327,000 inpatient hospital stays, 78,000 outpatient hospital visits, 195,000 emergency department visits, and 531,000 physician office visits in 1998.5 The cost of pancreatic diseases (direct and indirect costs) was estimated to be 2.5 billion dollars in the year 2000.5 In the same year, there were 2834 deaths in the United States from acute pancreatitis, making it the 14th most common cause of deaths due to gastrointestinal (GI) diseases.6 Acute pancreatitis ranks as the second most common inpatient GI diagnosis in the United States after cholelithiasis and acute cholecystitis and ahead of acute appendicitis.6
The incidence of acute pancreatitis appears to be increasing.7–9 As the population is becoming increasingly overweight, the incidence of gallstones, the most common cause of acute pancreatitis is rising. Unfortunately, during this same period, the overall mortality rate from acute pancreatitis has declined only gradually to approximately 5% to 10%.10–12
DEFINITIONS
Many pancreatologists use the 1992 Atlanta Symposium definition of acute pancreatitis,13 which is an acute inflammatory process of the pancreas with variable involvement of other regional tissues or remote organ systems. Acute pancreatitis is best defined clinically by a patient presenting with two of the following criteria12: (1) symptoms, such as epigastric pain, consistent with the disease; (2) a serum amylase or lipase greater than three times the upper limit of normal; or (3) radiologic imaging consistent with the diagnosis, usually using computed tomography (CT) or magnetic resonance imaging (MRI). Pancreatitis is classified as acute unless there are CT, MRI, or endoscopic retrograde cholangiopancreatography (ERCP) findings of chronic pancreatitis. Then pancreatitis is classified as chronic pancreatitis, and any episode of acute pancreatitis is considered an exacerbation of inflammation superimposed on chronic pancreatitis (see Chapter 59).
Once the diagnosis is established, patients are classified as having mild or severe pancreatitis. Mild acute pancreatitis consists of interstitial (edematous) pancreatitis on imaging, minimal or no extrapancreatic organ dysfunction, and typically an uneventful recovery. Severe pancreatitis manifests as organ failure or local complications such as necrosis, abscess, or pseudocyst. The Atlanta criteria13 (Table 58-1) defines severity by the presence of organ failure or pancreatic necrosis on dynamic contrast–enhanced CT scan (Figs. 58-1 and 58-2). Other acceptable markers of severe pancreatitis include three or more of Ranson’s 11 criteria for non-gallstone pancreatitis (Table 58-2),14 and an Acute Physiology and Chronic Health Evaluation (APACHE-II) score of greater than eight.15
Table 58-1 Atlanta Criteria for Severe Acute Pancreatitis13
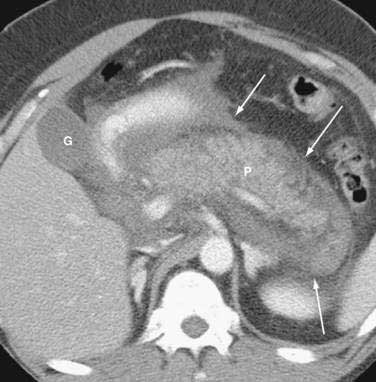
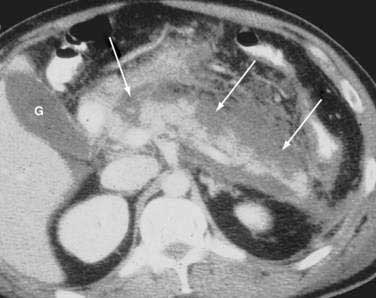
Table 58-2 Ranson’s Prognostic Criteria14,215
| NON-GALLSTONE PANCREATITIS (1974) | GALLSTONE PANCREATITIS (1982) |
|---|---|
| At Admission | |
| Age >55 yr | Age >70 yr |
| White blood cells >16,000/mm3 | >18,000/mm3 |
| Blood glucose >200 mg/dL | >220 mg/dL |
| Serum lactate dehydrogenase >350 IU/L | >400 IU/L |
| Serum aspartate aminotransferase >250 IU/L | >250 IU/L |
| During Initial 48 hr | |
| Hematocrit decrease of >10 % | >10% |
| Blood urea nitrogen increase of >5 mg/dL | >2 mg/dL |
| Serum calcium <8 mg/dL | <8 mg/dL |
| Arterial po2 <60 mm Hg | NA |
| Serum base deficit >4 mEq/L | >5 mEq/L |
| Fluid sequestration >6 L | >4 L |
NA, not applicable.
It is important to use precise terms in describing the anatomic complications of acute pancreatitis. The ability to apply appropriate therapy depends on a clear understanding of these terms. An old term that should be used only sparingly is phlegmon. Although this term is often used by radiologists to describe an inflammatory mass, this term has carried different meaning to gastroenterologists, internists, and surgeons. Whereas patients with interstitial pancreatitis have a normally perfused gland, manifesting contrast-enhanced CT as a normal bright appearance indicating flow throughout the gland, patients with necrotizing pancreatitis have greater than 30% of the gland not perfused, with low attenuation. Pancreatic necrosis consists of focal or diffuse nonviable pancreatic parenchyma and usually peripancreatic fat necrosis. Pancreatic necrosis can be sterile or infected. Peripancreatic necrosis describes necrotic fatty and stromal tissue around the pancreas. It is more important to surgeons because this is typically not appreciated on imaging. However, the presence of peripancreatic necrosis may delineate a more complicated course for patients with acute pancreatitis. An acute fluid collection is fluid located in or near the pancreas that lacks a definite wall and typically occurs early in the course of acute pancreatitis. On CT scan these collections appear as a low attenuation mass with poor margins and no capsule. It is very difficult to distinguish acute fluid collections in the pancreatic parenchyma from pancreatic necrosis. An acute fluid collection occurs in 30% to 50% of cases of acute pancreatitis and most resolve spontaneously.16 A pseudocyst is a fluid collection that persists for 4 to 6 weeks and becomes encapsulated by a wall of fibrous or granulation tissue. Pseudocysts are located adjacent to or off the body of the pancreas. At times these enzyme-rich fluid-filled sacks can be found distantly in the pelvis and chest. When a pseudocyst is located within the body of the pancreas, the cyst may contain necrotic pancreatic debris even when the pseudocyst is fluid-appearing with low attenuation on CT. The term for a walled-off fluid-appearing pseudocyst-like structure involving the pancreas is walled off pancreatic necrosis (WOPN). A pancreatic abscess is a circumscribed intra-abdominal collection of pus occurring after an episode of acute pancreatitis or pancreatic trauma. It usually develops close to the pancreas and contains little pancreatic necrosis. Due to confusion of whether an abscess represents an infected pseudocyst or infected pancreatic necrosis, the term abscess should be used sparingly. Because of important differences in management, it is best to use the terms infected pseudocyst and infected necrosis. The term hemorrhagic pancreatitis should also be used with caution, and this term is not a synonym for necrotizing pancreatitis. Hemorrhage is more commonly associated with pseudoaneurysm, an erosion of peripancreatic blood vessels with hemoperitoneum. Unfortunately, hemorrhagic pancreatitis has more commonly been used to inappropriately describe necrotizing pancreatitis.
Of all these terms, the most important distinction is that between pancreatic necrosis and pseudocyst. WOPN is pancreatic necrosis that has liquefied after five to six weeks.17 Similar to a pseudocyst, a wall develops. However, whereas a pseudocyst always contains fluid, pancreatic necrosis, even if walled off early, contains a significant amount of debris that only becomes liquefied after five to six weeks. No attempt should be made to drain WOPN early (less than four weeks) because the debris is typically thick, often with the consistency of rubber early in the course of the disease. After five to six weeks, WOPN can be treated similar to the fluid-filled pseudocyst and drained surgically, endoscopically, or percutaneously.
NATURAL HISTORY
Acute pancreatitis appears to have two distinct stages. The first stage is related to the pathophysiology of the inflammatory cascade. This first phase usually lasts a week. During this phase, the severity of acute pancreatitis is related to extrapancreatic organ failure secondary to the patient’s systemic inflammatory response elicited by acinar cell injury. Infectious complications are uncommon at this time. Fever, tachycardia, hypotension, respiratory distress, and leukocytosis are typically related to the systemic inflammatory response syndrome (SIRS). Multiple cytokines are involved, including platelet activating factor, tumor necrosis factor-α (TNF-α) and various interleukins (ILs) (see Chapter 2).
During the first week the initial state of inflammation evolves dynamically with variable degrees of pancreatic and peripancreatic ischemia or edema to either resolution or to irreversible necrosis and liquefaction, or the development of fluid collections in and around the pancreas. The extent of the pancreatic and peripancreatic changes is usually proportional to the severity of extrapancreatic organ failure. However, organ failure may develop independent of pancreatic necrosis.17
Approximately 75% to 80%, of patients with acute pancreatitis have a resolution of the disease process (interstitial pancreatitis) and do not enter the second phase. However, in 25% of patients, a more protracted course develops, often related to the necrotizing process (necrotizing pancreatitis) lasting weeks to months. The mortality peak in the second phase is related to a combination of factors, including organ failure secondary to sterile necrosis, infected necrosis, or complications from surgical intervention.11,12,18–20 There are two peaks for mortality. Most studies in the United States and Europe reveal that about half the deaths occur within the first week or two, usually of multiorgan failure.19–21 Death can be very rapid. About one quarter of all deaths in Scotland occurred within 24 hours of admission and one third within 48 hours.21 After the second week of illness, patients succumb to pancreatic infection associated with multiorgan failure. Some studies in Europe report a very high late mortality rate from infection.22 Patients who are older and have comorbid illnesses have a substantially higher mortality rate than younger healthier patients. In those who survive their illness, severe pancreatic necrosis can scar the pancreas, resulting in a stricture of the main pancreatic duct with subsequent obstructive chronic pancreatitis and permanent diabetes and malabsorption.23
PATHOGENESIS
The initial step in the pathogenesis of acute pancreatitis is conversion of trypsinogen to trypsin within acinar cells in sufficient quantities to overwhelm normal mechanisms to remove active trypsin (see Fig. 57-3). Trypsin, in turn, catalyzes conversion of proenzymes, including trypsinogen and inactive precursors of elastase, phospholipase A2 (PLA2), and carboxypeptidase, to active enzymes. Trypsin also may activate the complement and kinin systems. Active enzymes autodigest the pancreas and initiate a cycle of releasing more active enzymes. Normally small amounts of trypsinogen are spontaneously activated within the pancreas, but intrapancreatic mechanisms quickly remove activated trypsin. Pancreatic secretory trypsin inhibitor (PSTI, now called SPINK1) binds and inactivates about 20% of the trypsin activity. Other mechanisms for removing trypsin involve mesotrypsin, enzyme Y, and trypsin itself, which splits and inactivates trypsin. The pancreas also contains nonspecific antiproteases such as α1-antitrypsin and α2-macroglobulin. Additional protective mechanisms are the sequestration of pancreatic enzymes within intracellular compartments of the acinar cell during synthesis and transport and the separation of digestive enzymes from lysosomal hydrolases as they pass through the Golgi apparatus, which is important because cathepsin B activates trypsin from trypsinogen. Low intra-acinar calcium concentrations also prevent further autoactivation of trypsin.
In experimental pancreatitis, activation of trypsin occurs within 10 minutes, and large amounts of trypsin24 and increased concentrations of trypsinogen activation peptide (TAP) accumulate within the pancreas.25,26 TAP is cleaved when trypsinogen is activated to trypsin, and concentrations of TAP in plasma, urine, and ascites correlate with the severity of the pancreatic inflammatory response, with the highest levels associated with acinar necrosis and intrapancreatic hemorrhage.27,28
Co-localization of pancreatic enzymes in lysosomes, followed by acinar cell injury, is an attractive hypothesis for the pathogenesis of acute pancreatitis, but the relevance of co-localization to the pathogenesis of acute pancreatitis is unclear. Activation of trypsinogen occurs before biochemical or morphologic injury to acinar cells, in association with co-localization of lysosomal enzymes, such as cathepsin B, and digestive enzymes, including trypsinogen within unstable vacuoles.27,28 Complete inhibition of pancreatic cathepsin B activity in vitro prevents trypsinogen activation induced by the cholecystokinin (CCK) analog cerulein,29 supporting the co-localization hypothesis. Thus, complete inhibition of cathepsin B may prevent or be a treatment for acute pancreatitis. However, enzyme co-localization may occur without inducing significant acinar cell injury.30
Two other features of experimental acute pancreatitis are early blockade of the secretion of pancreatic enzymes while enzyme synthesis continues and disruption of the paracellular barrier of acinar cells and intralobular pancreatic duct cells. The disruption facilitates the extravasation of pancreatic enzymes from acinar cells and from the duct lumen into interstitial spaces. This phenomenon may explain the rapid development of interstitial edema and the increase of pancreatic enzymes in the serum.31
As discussed in Chapter 57, the discovery of genetic mutations associated with hereditary pancreatitis also lends support to the hypothesis that intrapancreatic activation of pancreatic zymogens is central to the pathogenesis of acute pancreatitis.32–35 The mutant trypsin in hereditary pancreatitis (usually R122H or N29I mutation) causes trypsin to be resistant to lysis or causes premature trypsinogen activation (gain of function mutation) leading to autodigestion of the pancreas and episodes of acute pancreatitis.36,37
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have also been implicated in pancreatitis (see Chapter 57). CFTR anion channel allows for chloride and bicarbonate secretion into the pancreatic ducts and thus allows flushing of the liberated enzymes and proenzymes into the duodenum. There are more than 1200 mutations that have been described for the CFTR gene. Some of these are considered severe and some mild. Homozygote severe mutations produce a viscid, concentrated, acidic pancreatic juice leading to ductal obstruction and pancreatic insufficiency in infanthood. Heterozygotes of minor or major mutations may lead to acute recurrent or chronic pancreatitis by altering acinar or ductal cell function (e.g., alteration of bicarbonate conductance).
A third genetic abnormality associated with pancreatitis is mutation of the SPINK1 gene.38 As noted, SPINK1 protects the pancreatic acinar cell by inhibiting prematurely activated trypsin. Mutations of this gene presumably limit the activity of this protein, but the exact mechanism is unclear.
The pathogenesis of gallstone-related pancreatitis is unknown (see Chapter 65). Factors that may initiate gallstone pancreatitis include reflux of bile into the pancreatic duct39,40 or obstruction of the pancreatic duct at the ampulla from stone(s) or edema resulting from the passage of a stone.41 Reflux of bile into the pancreatic duct could occur when the distal bile and pancreatic ducts form a common channel and a gallstone becomes impacted in the duodenal papilla. Alternatively, bile could reflux into the pancreatic duct from the duodenum through an incompetent sphincter of Oddi injured by recent passage of a gallstone.
Experimentally, reflux of bile into the pancreatic duct, particularly if infected or mixed with pancreatic enzymes, causes pancreatic injury. Mixtures of bile and pancreatic enzymes increase the permeability of the main pancreatic duct, which is associated with local parenchymal inflammation.42 The common channel theory is somewhat problematic because pancreatic duct pressure is invariably higher than bile duct pressure, making bile reflux into the pancreatic duct unlikely. Reflux of bile from the duodenum also is unlikely because pancreatitis does not occur in conditions with easily demonstrable reflux, such as after surgical sphincteroplasty or endoscopic sphincterotomy.
A popular opinion for the mechanism of gallstone pancreatitis is that an impacted gallstone in the distal bile duct obstructs the pancreatic duct, which increases pancreatic pressure, thereby damaging ductal and acinar cells. Experiments in the opossum that support this theory are the observations that ligation of the pancreatic duct causes severe necrotizing pancreatitis41 and that decompression of the ductal system within three days prevents progression to acinar cell necrosis and severe inflammation.43
PATHOPHYSIOLOGY
The release of pancreatic enzymes damages the vascular endothelium, the interstitium, and acinar cells.43–45 Acinar injury leads to expression of endothelial adhesion molecules (e.g., VCAM-1), which further propagates the inflammatory response.46 Microcirculatory changes, including vasoconstriction, capillary stasis, decreased oxygen saturation, and progressive ischemia, occur early in experimental acute pancreatitis. These abnormalities increase vascular permeability and lead to edema of the gland (edematous or interstitial pancreatitis). Vascular injury could lead to local microcirculatory failure and amplification of the pancreatic injury. It is uncertain whether ischemia-reperfusion injury occurs in the pancreas.40 Reperfusion of damaged pancreatic tissue could lead to the release of free radicals and inflammatory cytokines into the circulation, which could cause further injury. In early stages of animal and human pancreatitis, activation of complement and the subsequent release of C5a play significant roles in the recruitment of macrophages and polymorphonuclear leukocytes.47–49 Active granulocytes and macrophages release proinflammatory cytokines in response to transcription factors such as nuclear factor κB (NF-κB). Proinflammatory cytokines include TNF, IL-1, IL-6, and IL-8, and platelet-activating factor (PAF). Proinflammatory cytokines frequently are followed by anti-inflammatory cytokines (IL-2, IL-10, IL-11) that attempt to down-regulate inflammation.48 Other mediators of inflammation include arachidonic acid metabolites (prostaglandins, PAF, and leukotrienes), nitric oxide, proteolytic and lipolytic enzymes, and reactive oxygen metabolites that overwhelm scavenging by endogenous antioxidant systems. These substances also interact with the pancreatic microcirculation to increase vascular permeability, which induces thrombosis and hemorrhage and leads to pancreatic necrosis. A recent study suggests that gene polymorphisms that affect acinar cell glutathione concentrations may lead to increased oxidant stress and more severe pancreatitis.50 Meanwhile, ischemia and severe inflammation of the gland can lead to disruption of the main and secondary pancreatic ducts, leading to local fluid accumulations within and surrounding the pancreas that can eventuate into pseudocysts.51,52
Some patients with severe pancreatic damage develop systemic complications, including fever, acute respiratory distress syndrome (ARDS), pleural effusions, renal failure, shock, myocardial depression, and metabolic complications. SIRS is common in patients with acute pancreatitis and is probably mediated by activated pancreatic enzymes (phospholipase, elastase, trypsin) and cytokines (TNF, PAF) released into the portal circulation from the inflamed pancreas.53 Cytokines reaching the liver activate hepatic Kupffer cells, which, in turn, induces hepatic expression and secretion of cytokines into the systemic circulation. These cause acute phase protein synthesis (C-reactive protein [CRP], IL-6) and may cause SIRS and damage to the kidneys, lungs, and other organs leading to multiorgan dysfunction and failure.54
ARDS may be induced by active phospholipase A (lecithinase), which digests lecithin, a major component of lung surfactant. Acute renal failure has been explained on the basis of hypovolemia and hypotension. Myocardial depression and shock are likely secondary to vasoactive peptides and a myocardial depressant factor. Metabolic complications include hypocalcemia, hyperlipidemia, hyperglycemia with or without ketoacidosis, and hypoglycemia. The pathogenesis of hypocalcemia is multifactorial and includes hypoalbuminemia (the most important cause), hypomagnesemia, calcium-soap formation, hormonal imbalances (e.g., involving parathyroid hormone, calcitonin, and glucagon), binding of calcium by free fatty acid–albumin complexes, intracellular translocation of calcium, and systemic exposure to endotoxin.55
Pancreatic infection (infected necrosis and infected pseudocyst) can occur from the hematogenous route or from translocation of bacteria from the colon into the lymphatics. Under normal circumstances bacterial translocation does not occur because there are complex immunologic and morphologic barriers. However, during acute pancreatitis, these barriers break down, which can result in local and systemic infection.56 Penetration of the gut barrier by enteric bacteria is likely due to gut ischemia secondary to hypovolemia and pancreatitis-induced arteriovenous shunting in the gut.57 Indeed, in canine experimental pancreatitis, luminal Escherichia coli translocate to mesenteric lymph nodes and distant sites.58 In feline experimental pancreatitis, enclosing the colon in impermeable bags prevents translocation of bacteria from the colon to the pancreas.59
PREDISPOSING CONDITIONS
Many conditions predispose to acute pancreatitis to varying degrees (Table 58-3). This list will undoubtedly continue to grow, and the number of cases diagnosed as “idiopathic” will decrease as our understanding of the disease improves. Gallstones and chronic alcohol abuse account for 70% of acute pancreatitis in the United States.
Table 58-3 Conditions That Predispose to Acute Pancreatitis
OBSTRUCTION
Gallstones
The most common obstructive process leading to pancreatitis is gallstones, which cause approximately 40% of cases acute pancreatitis,60 although only 3% to 7% of patients with gallstones develop pancreatitis. Gallstone pancreatitis is more common in women than men because gallstones are more frequent in women.61 Acute pancreatitis occurs more frequently when stones are less than 5 mm in diameter (odds ratio, 4 to 5),62 because small stones are more likely than large stones to pass through the cystic duct and cause ampullary obstruction. Cholecystectomy and clearing the bile duct of stones prevents recurrence, confirming the cause-and-effect relationship.61
Biliary Sludge and Microlithiasis
Biliary sludge is a viscous suspension in gallbladder bile that may contain small (<3 mm) stones (i.e., microlithiasis).63 Because small stones can hide in biliary sludge, the two are commonly referred together as biliary sludge and microlithiasis. Biliary sludge is asymptomatic in most patients. It is usually composed of cholesterol monohydrate crystals or calcium bilirubinate granules.64 On ultrasonography, sludge produces a mobile, low-amplitude echo that does not produce an acoustic shadow and that layers in the most dependent part of the gallbladder.
Sludge may result from functional bile stasis, such as that associated with prolonged fasting or total parenteral nutrition, or from mechanical stasis such as occurs in distal bile duct obstruction. In addition, the cephalosporin antibiotic ceftriaxone can complex with bile to form a sludge within the biliary system when its solubility in bile is exceeded; this rarely causes stones65 and the sludge disappears after stopping the drug. Commonly, biliary sludge is associated with idiopathic acute pancreatitis. However, the association between biliary sludge and acute pancreatitis is unproved. There is no prospective, randomized study documenting that removing sludge or microcrystals by cholecystectomy prevents further attacks of pancreatitis. Nevertheless, results of two uncontrolled studies suggest that biliary sludge can lead to pancreatitis, and that cholecystectomy, papillotomy, or ursodeoxycholic acid therapy reduces recurrent attacks of acute pancreatitis.64,66 In these two studies, the incidence of biliary sludge in presumed idiopathic pancreatitis was 67% and 74%, respectively. However, other investigators have detected microlithiasis or sludge in less than 10% of patients with recurrent acute pancreatitis.67,68 Until prospective controlled studies clarify the proper treatment of sludge and microlithiasis, firm recommendations about therapy cannot be made. Choices include cholecystectomy, ursodeoxycholic acid therapy, endoscopic sphincterotomy, or watchful waiting.
Tumors
Tumors, presumably by obstructing the pancreatic duct, can cause recurrent acute pancreatitis especially in individuals older than age 40 (see Chapter 60). The most common tumor that presents in this manner is intraductal papillary mucinous neoplasm (IPMN).69 Pancreatic adenocarcinoma can also present as acute pancreatitis in a small percentage of patients.70 Metastases from other primary tumors (lung, breast) to the pancreas have also caused pancreatitis.71 Large adenomas of the major papilla can likewise occasionally be the cause of obstructive pancreatitis.
Other Obstructive Causes
Other obstructive conditions that are rarely associated with acute pancreatitis are conditions discussed elsewhere in this text and include choledochoceles,72 duodenal diverticula,73 annular pancreas,74 and parasites that obstruct the pancreatico-biliary system such as ascaris75 or clonorchis.76 Ascariasis obstructing the pancreatic duct represents the second most common cause of acute pancreatitis in Kashmir.60
ALCOHOL, OTHER TOXINS, AND DRUGS
Ethyl Alcohol
Alcohol causes at least 30% of cases of acute pancreatitis,77 and alcohol is the most common etiology of chronic pancreatitis in developed countries. Interestingly, only 10% of chronic alcoholic patients develop chronic pancreatitis. The classic teaching is that alcohol causes chronic pancreatitis, and that alcoholic patients who present with clinically acute pancreatitis have underlying chronic disease.16 However, a few patients with alcohol-induced acute pancreatitis by clinical criteria do not have or progress to chronic pancreatitis, even with continued alcohol abuse.77,78 By contrast, a small percentage of chronic alcoholic patients develop attacks of acute pancreatitis that are indistinguishable from other forms of acute pancreatitis, but eventually develop chronic pancreatitis after 10 to 20 years of alcohol abuse. Early in the course of the disease, when attacks occur, the diagnosis of underlying chronic pancreatitis is difficult without tissue specimens because the diagnosis of chronic pancreatitis is usually made after definite signs of chronic pancreatitis appear (e.g., pancreatic calcification, exocrine and endocrine insufficiency, or typical duct changes by CT or ERCP). Most of the models described suggest possible mechanisms of alcohol-related injury, including perturbations in exocrine function, changes in cellular lipid metabolism, induction of oxidative stress, and activation of stellate cells. However, the exact mechanism remains unclear and may be related to other factors.
Bordalo and colleagues79 first proposed that alcohol was directly toxic to the acinar cell through a change in cellular metabolism. Alcohol produces cytoplasmic lipid accumulation within the acinar cells, leading to fatty degeneration, cellular necrosis, and eventual widespread fibrosis. Fatty acid ethyl esters, by-products of pancreatic ethanol metabolism, may be the key factor in this “toxic metabolic” change. Bordalo and colleagues suggested that alcohol produces a stepwise progression from fatty accumulation to fibrosis (direct toxic effects on cellular metabolism). The main limitation to this toxic-metabolic theory of alcohol toxicity is the lack of proof of the steatopancreatitis precursor to fibrosis seen in liver disease.80
Sarles80 emphasized the duality of acute and chronic pancreatitis; they were separate diseases with distinct pathogenesis. Whereas acute pancreatitis can be precipitated in patients with gallstones immediately, alcoholism requires years of toxin exposure. Alcohol modulates exocrine function to increase the lithogenicity of pancreatic fluid, leading to the formation of protein plugs and stones. Chronic contact of the stones with the ductal cells produces ulceration and scarring, resulting in obstruction, stasis, and further stone formation. Eventually, atrophy and fibrosis develop as a result of this obstructive process. Several studies have provided mechanisms in which alcohol promote stone formation, including the known precipitation of GP-2 (a Tamm-Horsfall–like protein),81 increased secretion and viscosity of pancreatic juice, and hypersecretion of enzymes and lactoferrin. In addition to these ethanol-mediated perturbations in pancreatic exocrine function, specific proteins have been implicated in stone formation. Pancreatic stones consist of a calcium carbonate crystalline lattice interspersed within a gel-like matrix formed of multiple fibrillar proteins and polysaccharides.82
In contrast to the stone theory, which is based on the de novo development of fibrosis without acute pancreatitis, the necrosis-fibrosis hypothesis envisions the development of fibrosis from recurrent, perhaps subclinical, acute pancreatitis. Inflammation and necrosis from the initial episodes of acute pancreatitis produce scarring in the periductular areas and scarring leads to obstruction of the ductules leading to stasis within the duct and subsequent stone formation. Support for this theory comes from histopathologic studies that revealed mild perilobular fibrosis in resolving acute pancreatitis, with marked fibrosis with ductal distortion occurring later. It is thought that a stepwise progression occurs to fibrosis from recurrent episodes of acute pancreatitis. Support for this theory is seen in a clinical study by Ammann and colleagues.83 In this study, 254 patients were prospectively followed after the first episode of alcoholic pancreatitis. There was a direct correlation between the frequency and severity of attacks to the rate of progression to chronic pancreatitis.
Whitcomb and Schneider have proposed an interesting hypothesis for chronic pancreatitis which unifies and incorporates recent knowledge in an attempt to reconcile prior theories.84 In at-risk individuals, the pancreatic acinar cells are stimulated by alcohol. Fibrosis does not occur because a profibrotic cellular infiltrate is not yet present. A sentinel event occurs as trypsinogen is activated. This event results in a massive inflammatory response. Cytokines are then released and work with the activation of stellate cells in the late phase. The attraction and activation of stellate cells set the stage for the development of fibrosis. If the inciting factors are removed, then the pancreas returns to normal. If the inciting factor, alcohol, is not removed, the acinar cells continue to secrete cytokines in response to the oxidative stress and the stellate cells continue to be activated. This model, using a sentinel event also represents a time for disease modifying therapy, when such therapy becomes available.
Other Toxins
Methyl alcohol,85 organophosphorous insecticides,86 and the venom of the Trinidad scorpion87 have been reported to induce pancreatitis. The mechanism of the latter two is thought to be by hyperstimulation of the pancreas. Smoking increases the risk of alcoholic and idiopathic pancreatitis, but not gallstone pancreatitis.88
Drugs
Medications are an infrequent but an important cause of acute pancreatitis.89 More than 120 drugs have been implicated, mostly from anecdotal case reports. Many case reports suffer from a combination of inadequate criteria for the diagnosis of acute pancreatitis, failure to rule out more common causes, or a lack of a rechallenge with the medication. Drug-induced pancreatitis rarely is accompanied by clinical or laboratory evidence of a drug reaction, such as rash, lymphadenopathy, or eosinophilia. Although a positive rechallenge with a drug is the best evidence available for cause and effect, it is not proof. It is clear that many patients with idiopathic pancreatitis or microlithiasis have recurrent attacks of acute pancreatitis. Therefore, stopping and restarting a drug with recurrence of pancreatitis may be a coincidence and not cause and effect. Despite the lack of a rechallenge, a drug may be strongly suspected if there is a consistent latency among the case reports between initiating the drug and the onset of acute pancreatitis. Table 58-4 shows the drugs with the greatest evidence for causing acute pancreatitis, those with rechallenges or with a relatively predictable latency.89
Table 58-4 Drugs Associated with Acute Pancreatitis*
* Class 1 and class 2 drugs. For class 1 drugs: two or more case reports published, absence of other causes of acute pancreatitis, rechallenge documented in at least one report. For class 2 drugs: four or more case reports published, absence of other causes of acute pancreatitis, consistent latency in at least 75% of cases published.
From Badalov N, Baradarian R, Iswara K, et al. Drug induced acute pancreatitis: An evidence based approach. Clin Gastroenterol Hepatol 2007; 101:454-76.
METABOLIC DISORDERS
Hypertriglyceridemia
Hypertriglyceridemia is perhaps the third most common identifiable cause of pancreatitis after gallstones and alcoholism. Serum triglyceride concentrations greater than 1000 mg/dL (11 mmol/L) may precipitate attacks of acute pancreatitis. Patients may have lactescent (milky) serum owing to increased concentrations of chylomicrons.90 The pathogenesis of hypertriglyceridemic pancreatitis is unclear, but the release of free fatty acids by lipase may damage pancreatic acinar cells or capillary endothelium.91
Hypertriglyceridemia may cause up to 5% of cases of acute pancreatitis. The association between hypertriglyceridemia and acute pancreatitis is best defined in children with rare inherited disorders of lipoprotein metabolism and severe hypertriglyceridemia92,93 who develop acute pancreatitis in early childhood. These children are homozygous for lipoprotein lipase deficiency or, even less commonly, apoprotein-CII (APO-CII) deficiency. Acute pancreatitis develops in 35%, 15%, and 30% to 40% of patients with type I, IIb, and V hyperlipidemia, respectively. Lowering serum triglyceride levels to less than 200 mg/dL (2.2 mmol/L) can prevent pancreatitis.
Most adults with hyperchylomicronemia have a mild form of genetically inherited type I or type V hyperlipoproteinemia and an additional acquired condition known to raise serum lipids (e.g., alcohol abuse, obesity, diabetes mellitus, hypothyroidism, pregnancy, estrogen91 or tamoxifen therapy, glucocorticoid excess, nephrotic syndrome, thiazide or beta blocker therapy). Typically three types of patients develop hypertriglyceridemia-induced pancreatitis. The first is a poorly controlled diabetic patient with a history of hypertriglyceridemia. The second is an alcoholic patient with hypertriglyceridemia detected on hospital admission. The third (15% to 20%) is a nondiabetic, nonalcoholic, nonobese person who has drug- or diet-induced hypertriglyceridemia. Drug-induced disease is more likely to occur if there is a background of hypertriglyceridemia prior to drug exposure.
Most persons who abuse alcohol have moderate, but transient, elevations of serum triglyceride levels. This condition is likely an epiphenomenon and not the cause of their pancreatitis94 because alcohol itself not only damages the pancreas but also increases serum triglyceride concentrations in a dose-dependent manner. For example, serum triglyceride concentrations greater than 227 mg/dL (2.5 mmol/L) occurred in 10%, 14%, and 20% of people who drank three to five, six to eight, or nine or more drinks per day, respectively.95 Alcoholic patients with severe hyperlipidemia often have a coexisting primary genetic disorder of lipoprotein metabolism.
Hypercalcemia
Hypercalcemia, of any cause, is rarely associated with acute pancreatitis. Proposed mechanisms include deposition of calcium in the pancreatic duct and calcium activation of trypsinogen within the pancreatic parenchyma.96 The low incidence of pancreatitis in chronic hypercalcemia suggests that factors other than the serum calcium per se (e.g., acute elevations of serum calcium) are responsible for pancreatitis. Acute calcium infusion into rats leads to conversion of trypsinogen to trypsin, hyperamylasemia, and dose-dependent morphologic changes of acute pancreatitis such as edema and acinar cell necrosis.
Hypercalcemia due to hyperparathyroidism has been associated with pancreatitis. However, primary hyperparathyroidism causes less than 0.5% of all cases of acute pancreatitis, and the incidence of acute pancreatitis in patients with hyperparathyroidism varies from 0.4% to 1.5%.97 Rarely, pancreatitis occurs with other causes of hypercalcemia, including metastatic bone disease, total parenteral nutrition, sarcoidosis, vitamin D toxicity, and infusions of calcium in high doses perioperatively during cardiopulmonary bypass.
INFECTIONS
Many infectious agents may cause acute pancreatitis,76 but often published reports do not meet usual standards for the diagnosis of pancreatitis or the infection. Using modern criteria for diagnosis of pancreatitis, definite pancreatitis exists if there is surgical, autopsy, or radiologic evidence; probable pancreatitis exists if there is biochemical evidence (more than three times the elevation of serum lipase or amylase) plus characteristic symptoms; and possible pancreatitis exists if there is only asymptomatic biochemical evidence. The definite criterion for an infection causing pancreatitis is finding the organism in the pancreas or pancreatic duct by stain or culture. Probable criteria for infection are culture of the organism from pancreatic juice or blood or serologic evidence combined with a characteristic clinical or epidemiologic setting. The criterion for a possible infection is culture of the organism from other body sites or serologic evidence of infection.
Using these criteria, definite pancreatitis has been associated with viruses (mumps, coxsackievirus, hepatitis B, cytomegalovirus, varicella-zoster, herpes simplex, Epstein-Barr, hepatitis A, and hepatitis C); the vaccine that contains attenuated measles, mumps, and rubella (MMR); bacteria (Mycoplasma, Legionella, Leptospira, Salmonella, tuberculosis, and brucellosis); fungi (Aspergillus and Candida albicans); and parasites (Toxoplasma, Cryptosporidium, Ascaris, Clonorchis sinensis). C. sinensis and Ascaris cause pancreatitis by blocking the main pancreatic duct. In acquired immunodeficiency syndrome (AIDS), infectious agents causing acute pancreatitis include cytomegalovirus, Candida, Cryptococcus neoformans, Toxoplasma gondii, and possibly Mycobacterium avium complex.76
An infectious agent should be suspected of causing acute pancreatitis if the characteristic syndrome caused by the infectious agent is present because this occurs 70% of the time.76 Because an infectious agent may be found in the pancreas without pancreatitis, routine search for an infection in idiopathic pancreatitis is not recommended because false-positive test results may result. In addition, it is unknown whether treating an infectious agent reverses pancreatic pathology.
VASCULAR DISEASE
Rarely, pancreatic ischemia causes pancreatitis. In most cases it is mild, but fatal necrotizing pancreatitis may occur. Ischemia may result from vasculitis (systemic lupus erythematosus)98 and polyarteritis nodosa,99 atheromatous embolization of cholesterol plaques from the aorta to the pancreas after transabdominal angiography,100 intraoperative hypotension,101 hemorrhagic shock,102 ergotamine overdose, and transcatheter arterial embolization for hepatocellular carcinoma. Also, ischemia is one possible explanation for pancreatitis after cardiopulmonary bypass. In pigs, cardiogenic shock induced by pericardial tamponade causes vasospasm and selective pancreatic ischemia due to activation of the renin-angiotensin system.103 Acute pancreatitis has occurred in long-distance runners, which may be on an ischemic basis.104
TRAUMA
Either penetrating trauma (gunshot or stab wounds) or blunt trauma can damage the pancreas.105 In most cases there is also injury to adjacent viscera. Laparotomy is essential in all cases of penetrating trauma to assess and treat all intra-abdominal injuries, including those to the pancreas. Blunt trauma results from compression of the pancreas by the spine, such as in an automobile accident. In blunt trauma it is important to determine preoperatively whether there is injury to the pancreas because depending on the severity of pancreatic injury, it will be necessary to include the pancreas in the surgical plan. Secondly, even in the absence of serious injury to adjacent organs, surgery or endoscopic therapy may be necessary to treat a pancreatic ductal injury.
Diagnosis is highly dependent on CT, MRI, or magnetic resonance cholangiopancreatography (MRCP), which may show enlargement of a portion of the gland caused by a contusion or subcapsular hematoma, pancreatic inflammatory changes, or fluid within the anterior pararenal space if there is ductal disruption. The CT may be normal during the first two days despite significant pancreatic trauma. If there is a strong clinical suspicion of pancreatic injury or if the CT or MRCP scan shows an abnormality, ERCP is required to define whether there is pancreatic duct injury. If the pancreatic duct is intact and there are no other significant intra-abdominal injuries, surgery is not required. However, if ERCP reveals duct transection with extravasation of pancreatic fluid and there are no other intra-abdominal injuries, stenting of the pancreatic duct may be successful.106 Serious injuries to the pancreas can be treated with appropriate débridement. Associated injuries to the duodenum or bile duct can be treated by biliary diversion, gastrojejunostomy, and feeding jejunostomy. External pancreatic fistulas occur in approximately one third of patients after surgery for pancreatic trauma. Octreotide may be beneficial after pancreatic injury.107
POST-ERCP
Acute pancreatitis is the most common and feared complication of ERCP, associated with substantial morbidity and occasional mortality. About 500,000 ERCPs are performed annually in the United States. Asymptomatic hyperamylasemia occurs after 35% to 70% of ERCPs.108 Acute pancreatitis occurs in 5% of diagnostic ERCPs, 7% of therapeutic ERCPs, and up to 25% in those with suspected sphincter of Oddi dysfunction or in those with a history of post-ERCP pancreatitis.109 About half the cases are moderate to severe in intensity.
The mechanisms that lead to post-ERCP pancreatitis are complex and not fully understood. Rather than a single pathogenesis, post-ERCP pancreatitis is believed to be multifactorial, involving a combination of chemical, hydrostatic, enzymatic, mechanical, and thermal factors. Although there is some uncertainty in predicting which patients will develop acute pancreatitis following ERCP, a number of risk factors acting independently or in concert have been proposed as predictors of post-ERCP pancreatitis (Table 58-5).110–113 Identification of these risk factors for post-ERCP pancreatitis is essential to recognize cases in which ERCP should be avoided if possible, or in which protective endoscopic or pharmacologic interventions should be considered.
Table 58-5 Factors That Increase the Risk of Post-ERCP Pancreatitis
| Patient Related |
| Young age, female gender, suspected sphincter of Oddi dysfunction, recurrent pancreatitis, history of post-ERCP pancreatitis, normal serum bilirubin |
| Procedure Related |
| Pancreatic duct injection, difficult cannulation, pancreatic sphincterotomy, precut access, balloon dilation |
| Operator or Technical Related |
| Trainee (fellow) participation, nonuse of a guidewire for cannulation, nonuse of a pancreatic duct stent in high-risk procedures |
ERCP, endoscopic retrograde cholangiopancreatography.
In general, the more likely a patient is to have an abnormal bile duct or pancreatic duct, the less likely the patient will develop post-ERCP pancreatitis. Cheng created a 160 variable database that prospectively evaluated more than 1000 patients from 15 centers in the midwestern United States.112 Their study emphasized the role of patient factors, including age, sphincter of Oddi dysfunction (SOD), prior history of post-ERCP pancreatitis, and technical factors, including number of pancreatic duct (PD) injections, minor papilla sphincterotomy, and operator experience. The patient most at risk of developing post-ERCP pancreatitis was a woman with suspected choledocholithiasis and normal bilirubin, who underwent a sphincterotomy and no stone was found. In this patient population, more than a quarter of patients (27%) developed post-ERCP pancreatitis. MRCP and endoscopic ultrasound, which do not cause pancreatitis, can provide useful information (perhaps as accurate as ERCP) in many of these cases and are preferred modalities in the initial evaluation of such patients.
Early recognition of post-ERCP pancreatitis may be possible by evaluating serum amylase or lipase after the procedure.114,115 In a study that involved 231 patients, the two-hour serum amylase and lipase were more accurate than a clinical assessment in distinguishing nonpancreatitis abdominal pain from post-ERCP acute pancreatitis. Serum values greater than 276 IU/L for amylase and greater than 1000 IU/L for lipase obtained from serum two hours after the procedure had almost a 100% positive predictive value for post-ERCP pancreatitis.116 More recently, Ito and colleagues found that if the serum amylase was normal at three hours, only 1% of patients had post-ERCP pancreatitis compared with 39% if the amylase was greater than five times the upper limit of normal.117 A serum amylase or lipase alone should not guide a decision of whether a patient has post-ERCP pancreatitis. However, it appears that the tests may assist the clinician’s assessment of a patient with post-ERCP abdominal pain.
Although there has been an interest in developing medications that can prevent post-ERCP pancreatitis, studies have failed to identify a medication worthy of widespread use. In terms of attenuating the inflammatory response, the most promising results have been seen with nonsteroidal anti-inflammatory drugs (NSAIDs). Two clinical trials have been published evaluating the role of diclofenac in reducing the incidence of post-ERCP pancreatitis.116,118 Both trials placed patients on 100 mg of diclofenac by rectal suppository and both showed a reduction in the incidence of acute pancreatitis. However, another trial failed to show any benefit to diclofenec in preventing post-ERCP pancreatitis.119
It has been suggested that relaxation of the sphincter of Oddi following ERCP will promote pancreatic drainage and prevent acute pancreatitis. Several agents have been used in the effort to relax the sphincter of Oddi with the purpose of preventing post-ERCP pancreatitis. There have been three placebo-controlled randomized studies evaluating the use of nitroglycerin during ERCP, with negative results.120–122 Furthermore, trials of oral nifedipine,123,124 sprayed lidocaine,125 and injected botulinum toxin126 failed to demonstrated any benefit in the reduction of severity or incidence of post-ERCP pancreatitis. There have been several studies evaluating the role of glucocorticoids using a variety of agents including oral prednisolone or intravenous hydrocortisone or methylprednisolone, with essentially no benefit in reduction of severity or incidence of post-ERCP pancreatitis.
Gabexate is a protease inhibitor with anti-inflammatory properties. The in vivo effect of gabexate on inhibiting circulating trypsin is greater than most other protease inhibitors. In 1995, Messori and colleagues127 published a meta-analysis of five trials128–132 showing a statistically significant reduction in the incidence of complications in patients receiving gabexate after the development of post-ERCP pancreatitis. However, the trials were small and had a limited number of patients. Several additional trials have been published with conflicting results.133–137 Although data are conflicting, it appears that infusions of the drug would likely need to be started 1 to 2 hours pre-ERCP and continued for 12 hours following ERCP to show a beneficial effect of the drug. In patients with a low risk, the costs likely outweigh any benefit. Currently, gabexate is not available in the United States.
Theoretically, inhibition of exocrine pancreatic secretion could prevent post-ERCP pancreatitis by inducing “rest” to a damaged gland. Although an attractive concept, there is little scientific basis to support this approach. Somatostatin and its synthetic octapeptide analog, octreotide, are potent inhibitors of pancreatic secretion. Although several trials of somatostatin have demonstrated an efficacy in reducing the incidence of post-ERCP pancreatitis,138–143 the majority of the studies do not support the routine use of this medication.144–150 Octreotide, the analog of somatostatin,151 was only effective in reducing post-ERCP hyperamylasemia, and did not reduce the incidence of post-ERCP pancreatitis.
Pancreatic stent placement clearly decreases the risk of post-ERCP pancreatitis in high-risk patients.152 Placement of pancreatic duct stents has become a standard practice for patients who are thought to be at high risk for pancreatitis after the procedure (see Table 58-5).153 Pancreatic duct stent placement is effective presumably by preventing cannulation-induced edema that can cause pancreatic duct obstruction. Pancreatic sphincter hypertension is believed to be an important causative factor in post-ERCP pancreatitis and may explain the high risk of pancreatitis in patients with SOD. There is prolonged alleviation of ductal obstruction when pancreatic stents are placed. Typically, 3- to 5-French unflanged pancreatic stents are used in the following settings: SOD, difficult cannulation, biliary orifice balloon dilation, and precut sphincterotomy. In general, pancreatic duct stents are not beneficial in patients who undergo routine biliary sphincterotomy. In all reported studies, which cumulatively include 1500 high-risk patients undergoing ERCP, only 1 patient developed severe pancreatitis after a pancreatic duct stent had been placed.154 Aside from the obvious benefits in preventing post-ERCP pancreatitis regarding morbidity and mortality, prophylactic stent placement is a cost-effective strategy for the prevention of post-ERCP pancreatitis for high-risk patients.155
Guidewire cannulation, whereby the biliary or pancreatic duct is initially cannulated by a guidewire inserted through the catheter or sphincterotome, has been shown to decrease the risk of pancreatitis (see Table 58-5).156 In a study of 400 consecutive patients who underwent ERCP by a single endoscopist, randomized to initial cannulation with contrast versus initial cannulation by a guidewire under fluoroscopic control, pancreatitis rates were profoundly different. No cases of acute pancreatitis were seen in the guidewire group compared with eight cases in the standard contrast group (P < 0.001). Cannulation success rates between the standard contrast and guidewire techniques were comparable 98.5% versus 97.5%. A later study157 confirmed a decrease in post-ERCP pancreatitis in 300 patients prospectively randomized to guidewire cannulation compared with conventional radiocontrast. However, the decrease in post-ERCP pancreatitis appears to be related to a decreased need for precut sphincterotomy in patients undergoing guidewire cannulation.
POSTOPERATIVE
Postoperative pancreatitis can occur after abdominal or thoracic surgery.158 Pancreatitis occurs after 0.4% to 7.6% of cardiopulmonary bypass operations113,159 and after 6% of liver transplantations.160 Twenty-seven percent of patients undergoing cardiac surgery develop hyperamylasemia, and 1% develop necrotizing pancreatitis.113 Significant risks for pancreatitis after cardiopulmonary bypass are preoperative renal insufficiency, postoperative hypotension, and administration of calcium chloride perioperatively. Mortality from postoperative pancreatitis is said to be higher (up to 35%) than for other forms of pancreatitis. Contributors to morbidity and mortality from postoperative pancreatitis are delay in diagnosis, hypotension, medications (e.g., azathioprine/perioperative calcium chloride administration), and infections.
HEREDITARY AND GENETIC
Hereditary pancreatitis is an autosomal dominant disorder with variable penetrance.161–169 It is discussed in Chapter 57.
CONTROVERSIAL CAUSES
Pancreas Divisum
Pancreas divisum is the most common congenital malformation of the pancreas occurring in 5% to 10% of the general healthy population (see Chapter 55). Controversy continues to surround the issue as to whether pancreas divisum with otherwise normal ductular anatomy is a cause of acute recurrent pancreatitis. The presumed mechanism of action in those who develop pancreatitis is that there is relative obstruction to the flow of pancreatic juice through the minor papilla. The arguments in favor of attributing pancreatitis to pancreas divisum are the following: (1) various series from ERCP referral centers show that patients referred with recurrent acute pancreatitis have a higher frequency of pancreas divisum than would be expected from the general population170; (2) multiple observational series report that performing endoscopic sphincterotomy or placing a stent across the minor papilla reduces the rate of recurrent pancreatitis156; and (3) there is one randomized controlled study suggesting that patients with pancreas divisum who are stented for one year have a lower frequency of attacks of pancreatitis than those not stented.171 The arguments against the association are the following: (1) there are studies to show that the rate of pancreatitis in pancreas divisum patients is the same as the general population172; (2) the observational reports are flawed in that follow-up was not long enough (usually only 1 to 2 years) and that recurrent acute pancreatitis is a disease of great variability163; (3) the single randomized study163 was flawed in that it was not blinded, was small (19 patients total), and its patients probably had chronic pancreatitis in that they had multiple pain attacks in between attacks of acute pancreatitis; (4) the risk of endoscopic therapy is considerable with a high rate of post-ERCP pancreatitis in patients with pancreas divisum,111,112 therefore, making the risk-benefit ratio of treating pancreas divisum endoscopically questionable; and (5) the rate of genetic abnormalities in patients with pancreas divisum and acute recurrent pancreatitis are either the same173 or higher161 than expected in the general population or population of patients with acute pancreatitis of other etiologies, suggesting a possible genetic source. For example, there appears to be a higher incidence of CFTR mutations in patients with pancreas divisum who develop acute pancreatitis.161 Therefore, it may not be the presence of pancreas divisum alone that predisposes to acute pancreatitis but other factors may be necessary to precipitate an attack.163









