CHAPTER 93 Acute Liver Failure
DEFINITION
Acute liver failure is defined as the rapid development of hepatocellular dysfunction, specifically coagulopathy and mental status changes (encephalopathy) in a patient without known prior liver disease.1 Acute liver failure is a clinical syndrome that represents the final common pathway of severe liver injury resulting from numerous infectious, immunologic, metabolic, vascular, and infiltrative disorders. The mechanism of liver injury in acute liver failure is most often severe hepatocellular necrosis, as occurs with acetaminophen toxicity or viral hepatitis. Acute liver failure can also result from severe cellular or mitochondrial dysfunction, as occurs with some forms of drug toxicity (e.g., antiretroviral agents), Wilson disease, and acute fatty liver of pregnancy.2
Acute liver failure (or fulminant hepatic failure) originally was defined by an interval of eight weeks or less between the onset of illness and appearance of encephalopathy.3 In an attempt to improve the prediction of the prognosis and outcome, O’Grady and colleagues divided patients into three groups based on the time interval between the onset of jaundice and encephalopathy: those with hyperacute liver failure (seven days or less), those with acute liver failure (eight to 28 days), and those with subacute liver failure (four to 24 weeks).4 In general, patients with hyperacute liver failure are more likely to develop cerebral edema and to recover without liver transplantation. By contrast, patients with subacute or late-onset hepatic failure are more likely to present with evidence of portal hypertension such as ascites and to have a low rate of survival without transplantation.5–7 Although the duration of illness may help predict prognosis, the overlap among patients with acute liver failure with varying presentations is great, and the duration of symptoms is largely related to the cause of liver failure. The original definition of acute liver failure (encephalopathy and coagulopathy within eight weeks of the illness onset) is used in this chapter, because this definition is the most widely used in clinical studies and in criteria for liver transplantation in the United States.
CAUSES
The underlying cause of acute liver failure in an individual patient is established by the patient’s history, serologic and molecular diagnostic test results, and characteristic radiologic or histologic features. The predominant cause of acute liver failure differs markedly throughout the world. In the United States and other Western countries, medications, including acetaminophen, and idiosyncratic drug toxicity are the most commonly identified causes of acute liver failure (Table 93-1).8,9 In France, Japan, and India, severe acute HBV infection is a leading cause of acute liver failure.10–12 In addition to these causes, numerous other, often rare, conditions can lead to acute liver failure (Table 93-2).
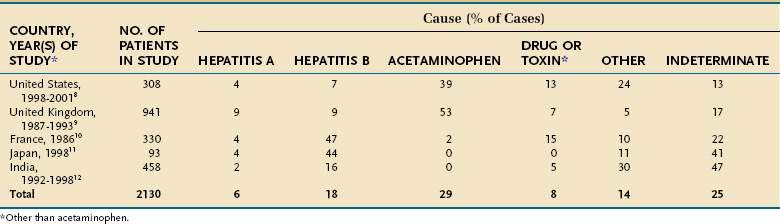
Table 93-2 Uncommon Causes of Acute Liver Failure
ACETAMINOPHEN TOXICITY
Acetaminophen is a dose-dependent hepatotoxin that, when ingested in excessive doses, can lead to life-threatening liver injury characterized by hypoprothrombinemia, towering aminotransferase elevations, and a normal or minimally elevated serum bilirubin level (Table 93-3). Measurement of serum acetaminophen levels is helpful in assessing the risk of hepatotoxicity following an acute overdose, but false-positive detection of acetaminophen in serum has been reported in some patients with deep jaundice if a colorimetric assay for acetaminophen is used.13 Because of the widespread availability of acetaminophen, intentional acetaminophen overdose (i.e., >10 g) is a common mode of attempted suicide, with over 60,000 cases reported each year in the United States.14 Although most patients who take an intentional overdose of acetaminophen recover, acute liver failure develops in a minority, and acetaminophen toxicity has become the leading cause of acute liver failure in both the United States and United Kingdom (Fig. 93-1).8,9 An increasing frequency of cases of unintentional acetaminophen overdose leading to acute liver failure has also been reported since the 1990s.15–17 In many such “therapeutic misadventures,” patients have ingested over-the-counter products containing acetaminophen as well as narcotic-acetaminophen congeners prescribed for an acute medical condition. Almost 10% of U.S. patients discharged from urban emergency rooms are given a prescription for an acetaminophen-narcotic congener, but most do not receive proper instruction regarding the need to reduce the dose or discontinue the use of other acetaminophen-based analgesics.18 Chronic heavy alcohol consumption may lower the threshold for acetaminophen toxicity in some patients by inducing cytochrome P450 enzyme activity (see Chapter 86).19 In addition, preexisting hepatic dysfunction and resulting glutathione depletion may predispose some patients to acetaminophen toxicity.20 Most patients with an unintentional overdose of acetaminophen have ingested large doses of acetaminophen-containing products over several days, but one study has suggested that ingestion of only 4 g of acetaminophen daily can lead to mild, transient serum aminotransferase elevations in 40% of healthy volunteers.21
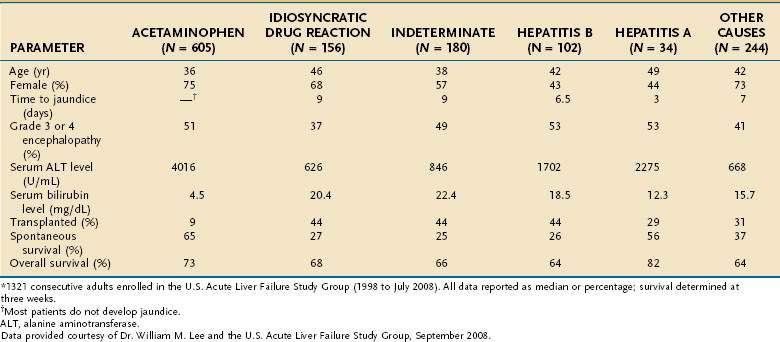
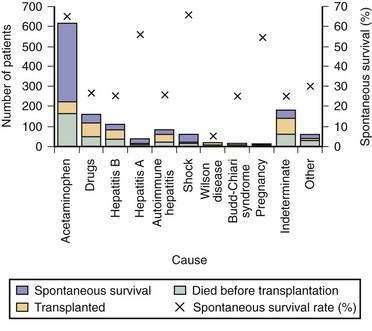
In the United Kingdom, restrictions on the quantity of acetaminophen dispensed, as well as blister packaging of products, were introduced in 1998 to reduce the incidence of acetaminophen toxicity. Since then, rates of hospitalization, acute liver failure, and the need for liver transplantation for acetaminophen toxicity have declined.22,23 In the United States, black box warnings on acetaminophen products and package labeling have been instituted, but the impact of these measures on the incidence and severity of acute liver failure caused by acetaminophen has not been evaluated. Additional measures that may prove useful include imposing limits on the number of tablets sold at one time, unbundling or reducing the dose of acetaminophen in prescription-narcotic congeners, and placing stronger warnings on package labeling.24,25
IDIOSYNCRATIC DRUG TOXICITY
Numerous prescription drugs, including various antibiotics, nonsteroidal anti-inflammatory drugs, and antiseizure medications, have been implicated in acute liver failure (see Chapter 86).26,27 In most cases, drug-induced acute liver failure is a rare and unpredictable event resulting from a metabolic idiosyncrasy that occurs in 1 in 10,000 to 1,000,000 patient–exposure years. Patients with drug-induced acute liver failure are frequently female (70%) and develop jaundice within six months of starting the suspected agent.8,26 Among 141 U.S. liver transplant recipients with drug-induced acute liver failure, isoniazid (16%), propylthiouracil (9%), phenytoin (7%), and valproic acid (7%) were the most commonly identified causative medications.28 Various over-the-counter herbal products and dietary supplements also have been associated with acute liver failure, including kava kava, weight loss supplements, and ephedra.29–31 In addition, severe hepatotoxicity has been associated with extracts of green tea, black cohosh, and adulterated traditional Chinese medicines (see Chapter 87).32–34 Unfortunately, herbal products and dietary supplements are not closely regulated during development, manufacturing, or marketing, and, in many mixtures, the hepatotoxic ingredient(s) cannot be identified.
Establishing a diagnosis of drug-induced acute liver failure is usually difficult because of the lack of specific laboratory markers, inability to rechallenge the patient, and limitations of available causality assessment instruments.35 In addition, less than 10% of patients have evidence of a hypersensitivity reaction associated with a rash or eosinophilia at presentation. The Drug-Induced Liver Injury Network (DILIN) was established to improve our understanding of the risk factors, mechanisms, and outcomes of drug-induced liver injury in the United States.36 The development of an evidence-based causality assessment instrument to assist with the early recognition and diagnosis of idiosyncratic drug-induced liver injury is a priority (see http://dilin.dcri.duke.edu for additional information). The primary treatment of drug-induced acute liver failure is to withdraw the culprit drug immediately once drug-induced liver injury is suspected. In selected patients with severe hepatotoxicity caused by α-methyldopa, nitrofurantoin, or minocycline and autoimmune features (i.e., hypergammaglobulinemia, autoantibodies, plasma cell infiltration on a liver biopsy specimen), treatment with glucocorticoids may be of benefit, but controlled studies are lacking.37,38 Overall, the outcome of drug-induced acute liver failure is poor, with a spontaneous survival rate of only 2% to 40% unless liver transplantation is performed emergently.
VIRAL INFECTIONS
Hepatotropic Viruses
Hepatitis A virus (HAV) and HBV infections are major causes of acute liver failure in many parts of the world, including India and other developing countries (see Table 93-1 and Chapters 77 and 78). Acute infection with HAV rarely leads to acute liver failure (<0.01% of cases), and, when it does, the prognosis is relatively good (see Fig. 93-1). Data from the U.S. Acute Liver Failure Study Group have indicated that the incidence of fulminant HAV infection is declining in parallel with the declining incidence of acute HAV in the general population because of increased immunization against HAV.39 Although HBV is the most common viral cause of acute liver failure, acute liver failure is an uncommon manifestation of acute HBV infection. Infection with hepatitis D virus (HDV), which requires coinfection with HBV, accounts for 4% of cases of acute liver failure in areas endemic for HBV.40 Older studies have suggested that virologic factors, including infection with precore or core promoter variants of HBV, may account for the development of acute liver failure in some patients.41 Subsequent studies have failed to demonstrate unique HBV mutants or variants associated with acute liver failure; the role of HBV genotypes in acute liver failure requires further investigation.42,43 Acute hepatitis E virus (HEV) infection is a leading cause of acute liver failure in India and other tropical countries but is rarely seen in Western countries (see Chapter 80).12,44 Pregnant women may be particularly prone to develop acute liver failure caused by HEV. European series have suggested that organ transplant recipients may be at increased risk of acquiring acute HEV infection and may develop chronic HEV infection.45,46 Hepatitis C virus (HCV) rarely causes acute liver failure.
Other Viruses
Nonhepatotropic viruses, including Epstein-Barr virus (EBV), cytomegalovirus (CMV), varicella-zoster virus, herpes simplex virus (HSV), and parvovirus B-19 infection account for less than 1% of cases of acute liver failure in adults.47,48 Whether these rare causes of acute liver failure are the result of viral variants or an aberrant host immune response to the virus is unclear. Making a diagnosis of acute liver failure caused by a nonhepatotropic virus is often difficult and frequently requires histologic confirmation, as well as polymerase chain reaction testing for viral deoxyribonucleic acid (DNA) in the serum. Almost 50% of patients with HSV-related acute liver failure have no characteristic skin lesions at presentation; the mortality rate is high because the diagnosis is usually delayed.49 Severe acute EBV, CMV, and HSV infection should be considered as possible causes of acute liver failure, particularly in immunosuppressed patients, because they can be treated successfully with antiviral therapy (see Chapter 81).
MISCELLANEOUS CAUSES
Acute liver failure occasionally develops in pregnant women, particularly during the third trimester (see Table 93-2).50 Acute fatty liver of pregnancy occurs in 0.0008% of all pregnancies and is associated with preeclampsia in over 50% of cases (see Chapter 38).51 Some affected women have an inherited deficiency in a fatty acid oxidation enzyme that can be identified by genetic testing. Wilson disease, a rare autosomal recessive disorder characterized by impaired biliary excretion of copper, can present as acute liver failure in up to 25% of cases (see Chapter 75). Most of these patients present in the second or third decade of life and have prominent hemolysis, a low serum alkaline phosphatase level, an elevated serum aspartate aminotransferase (AST)–to–alanine aminotransferase (ALT) ratio, increased urinary copper excretion, and Kayser-Fleischer rings.52 Prompt recognition and listing for liver transplantation are essential, because the outcome is otherwise fatal. Infrequent causes of acute liver failure include mushroom (Amanita phalloides) poisoning (see Chapter 87), Budd-Chiari syndrome (see Chapter 83), autoimmune hepatitis (see Chapter 88), and malignant infiltration of the liver (see Chapter 35). All the miscellaneous causes of acute liver failure combined account for 5% to 30% of cases of acute liver failure (see Table 93-1).
INDETERMINATE ACUTE LIVER FAILURE
Acute liver failure of unknown cause, defined by negative serologic testing for hepatitis A, B, C, D, and E and the absence of other known causes, constitutes 15% to 44% of cases of acute liver failure worldwide (see Table 93-1). Because many of these patients present with a viral prodrome, the hope has been that new, more sensitive molecular laboratory methods would identify a viral cause of acute liver failure of unknown cause. Occult HBV infection has been identified in the sera or livers of some patients with acute liver failure of unknown cause by some41,53 but not other42,43 investigators. Although HCV has been implicated as a cause of acute liver failure in a few patients, it is an exceedingly rare cause of acute liver failure in Western countries.53–55 Togavirus-like particles have been identified by electron microscopy in 7 of 18 liver explants from patients who underwent transplantation for indeterminate acute liver failure but are unlikely to be responsible for a substantial portion of cryptogenic cases of acute liver failure.56 The transfusion-transmitted virus (TTV) was found in the sera of patients with acute liver failure in initial studies, but TTV infection is not thought to be pathogenic.57,58 Studies have failed to demonstrate a link between hepatitis G virus (GB agent, or GBV-C), parvovirus B19, or SEN virus and indeterminate acute liver failure (see Chapter 81).59–61
Using a highly sensitive and specific assay for serum acetaminophen-cysteine adducts, Davern and colleagues identified adducts in 7 of 36 (19%) patients presumed to have indeterminate acute liver failure; the adduct levels were similar to those in patients with known acetaminophen hepatotoxicity.62 These patients tended to have high serum aminotransferase levels and low serum bilirubin levels at presentation, features similar to those seen in patients with a known acetaminophen overdose (see Table 93-3). Whether acetaminophen was the primary cause of liver injury or a cofactor requires further study.63 Regardless, patients with indeterminate acute liver failure should be evaluated rapidly for liver transplantation because of the low likelihood of spontaneous recovery (see later).
CLINICAL FEATURES
The clinical features of acute liver failure may result from the loss of critical hepatocellular functions (e.g., protein synthesis, intermediary metabolism, detoxification) and from effects on organs other than the liver. The major complications of acute liver failure, as well as their pathogenesis and medical management, are outlined in Table 93-4. The initial presentation usually includes nonspecific complaints such as nausea, vomiting, and malaise, and jaundice usually develops soon after. Hepatocellular injury leads to impaired elimination of bilirubin, depressed synthesis of coagulation factors I, II, V, VII, IX, and X, and diminished synthesis of glucose. In addition, decreased uptake and increased generation of intracellular lactate occur as a result of anaerobic glycolysis. These derangements manifest clinically as jaundice, coagulopathy, hypoglycemia, and metabolic acidosis. In addition to portending liver failure, coagulopathy increases the risk of gastrointestinal and intracranial hemorrhage, hypoglycemia can contribute to brain injury, and acidosis can contribute to hypotension.
Table 93-4 Pathogenesis and Management of Major Complications of Acute Liver Failure
| COMPLICATION | PATHOGENESIS | MANAGEMENT |
|---|---|---|
| Hypoglycemia | Diminished hepatic glucose synthesis | Blood glucose monitoring |
| Intravenous glucose supplementation (10% or 20% dextrose) | ||
| Encephalopathy | Cerebral edema | CT scan (if advanced encephalopathy) |
| ICP monitoring (if stage 3 or 4 encephalopathy) | Elevate head of the bed > 30 degrees | |
| Consider osmotherapy (mannitol) or barbiturates | ||
| Treat other contributing factors (e.g., hypoglycemia, hypoxemia, fever) | ||
| Reduce fever (cooling blankets, antibiotics) | ||
| Avoid benzodiazepines and other sedative medications | ||
| ? Moderate hypothermia (see text) | ||
| Infections | Reduced immune function | Aseptic medical, nursing care |
| Invasive procedures | Daily surveillance cultures of blood, urine, and sputum | |
| High index of suspicion for bacterial and fungal infection | ||
| Preemptive antibiotics for gram-negative organisms, anaerobes, and skin flora | ||
| Consider antifungal therapy if patient worsens despite antibacterial coverage | ||
| Gastrointestinal hemorrhage | Stress ulceration | Nasogastric tube placement |
| Intravenous H2 receptor antagonist or proton pump inhibitor | ||
| Coagulopathy | Reduced clotting factor synthesis | Parenteral vitamin K |
| Thrombocytopenia | Platelet infusions for bleeding and before procedures | |
| Fibrinolysis | Plasma infusions for bleeding and before procedures | |
| Cryoprecipitate for bleeding with hypofibrinogenemia | ||
| Recombinant factor VIIa (?) (see text) | ||
| Hypotension | Hypovolemia | Hemodynamic monitoring of central venous pressures |
| Decreased vascular resistance | Volume repletion with blood or colloid | |
| α-Adrenergic agents | ||
| Respiratory failure | ARDS (DAD) | Hemodynamic monitoring of central venous pressures |
| Mechanical ventilation | ||
| Pancreatitis | ?Hypoxia | Supportive care, including supplemental oxygen if needed |
| Abdominal CT to exclude necrotizing pancreatitis | ||
| Renal failure | Hypovolemia | Hemodynamic monitoring of central venous pressures |
| Hepatorenal syndrome | Volume repletion with blood or colloid | |
| Acute tubular necrosis | Avoidance of nephrotoxic agents (e.g., aminoglycosides, NSAIDs, contrast dye) | |
| Oral N-acetylcysteine prior to intravenous contrast agent | ||
| Hemofiltration, dialysis |
ARDS, acute respiratory distress syndrome; CT, computed tomography; DAD, diffuse alveolar damage; ICP, intracranial pressure; NSAIDs, nonsteroidal anti-inflammatory drugs.
HEPATIC ENCEPHALOPATHY AND CEREBRAL EDEMA
Hepatic encephalopathy is a defining criterion for acute liver failure. Encephalopathy in acute liver failure is thought to arise primarily from the development of cerebral edema and resulting intracranial hypertension, rather than from portosystemic shunting of toxins. In addition to cerebral edema, many of the other complications of acute liver failure, including hypoglycemia, sepsis, fever, hypoxemia, and hypotension, may contribute to neurologic abnormalities. The staging of encephalopathy in acute liver failure is similar to that used for patients with cirrhosis (see Chapter 92). In stage 1, patients have subtle changes in affect, altered sleep patterns, or difficulties with concentration. Stage 2 is characterized by drowsiness, disorientation, and confusion. Stage 3 is marked by somnolence and incoherence. In stage 4, frank coma with minimal (4A) or no (4B) response to noxious stimuli is detected. On physical examination, many patients have asterixis or a tremor in stage 1 or 2, whereas hyperreflexia, clonus, and muscular rigidity are common in stages 3 and 4. Although worrisome, these upper motor neuron signs do not portend a poor prognosis and can reverse with recovery or replacement of the failing liver.
Cerebral edema is found in up to 80% of patients who die of acute liver failure and is almost universal in patients with coma.64 The pathogenesis of cerebral edema in acute liver failure is poorly understood. It has been proposed to result, in part, from the actions of intestine-derived neurotoxins that escape hepatic clearance and are released into the systemic circulation.65 The demonstration of swollen endothelial and astroglial cells in the brains of patients with acute liver failure suggests a potential role for cytotoxic edema, possibly resulting from increased brain glutamine levels. On the other hand, vacuolization in the basement membranes of capillaries, consistent with disruption of the blood-brain barrier, suggests a vasogenic mechanism of cerebral edema in acute liver failure. In any event, an increased production of glutamine in the central nervous system as a result of high circulating levels of ammonia and intracerebral lactate is believed to be critical to the pathogenesis of cerebral edema. In one study, arterial ammonia levels were associated with the risk of uncal herniation and death in patients with acute liver failure.66 Another study suggested that a lack of reduction in arterial ammonia levels over time was associated with an increased risk of progression to cerebral edema.67
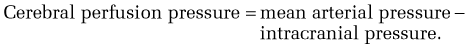
COAGULOPATHY AND BLEEDING
The liver is the major site of synthesis of coagulation factor and related inhibitory proteins. The reticuloendothelial cell system of the liver is involved in the clearance of activated clotting factors and their degradation products. Therefore, patients with acute liver failure frequently have a multifactorial coagulopathy and a resulting increased risk of bleeding and clotting. Laboratory features of fibrinolysis, hypofibrinogenemia, dysfibrinogenemia, and DIC are frequent in patients with acute liver failure.68 Thrombocytopenia also develops in most patients with acute liver failure and may be the result of increased destruction of platelets from a consumptive coagulopathy or reduced thrombopoietin production, bone marrow dysfunction, or the effects of medications.69 Clinically significant bleeding has been reported to occur in 10% to 20% of patients with acute liver failure; the most common sources are the upper gastrointestinal tract, nasopharynx, and skin puncture sites. Critically ill patients with acute liver failure have a particular propensity for gastrointestinal bleeding caused by acute portal hypertension, increased intracranial pressure, and coagulopathy.70









