Timothy A. Morris
The principle function of the lungs is gas exchange; hypercapnia indicates severe compromise of this vital function. Although disease may substantially affect any of the functional elements of the respiratory system, the term acute respiratory failure is used only when gas exchange is so severely impaired that arterial hypoxemia or hypercapnia occurs. Hypoxemia may involve a multitude of respiratory and metabolic processes and may occur in the absence of hypercapnia. However, hypercapnia is more directly linked to inadequate gas exchange and dysfunction of one or more elements of the respiratory system (e.g., control of breathing, mechanical performance of the lungs, respiratory muscle function, lung parenchyma, and vasculature). Hypercapnic respiratory failure is often referred to as alveolar hypoventilation and nearly always involves some level of hypoxemia as well. Specific values of arterial Pco2 (Paco2) that indicate hypercapnic respiratory failure are not well defined, but most experts agree that a Paco2 greater than 45 mmHg (in a previously eucapnic patient) reflects acute respiratory failure. In patients with chronic hypercapnic lung disease, a sudden increase of 5 mmHg or more of Paco2 from a previously stable level represents acute hypercapnic respiratory failure superimposed on chronic respiratory failure.
DIAGNOSIS
The hallmark of acute hypercapnic respiratory failure is an elevated Paco2. A rise in Paco2 signals that pulmonary “clearance” of carbon dioxide is inadequate; that is, more carbon dioxide is being produced by body metabolism than what the respiratory apparatus can clear by ventilation. This relationship is defined by the equation:
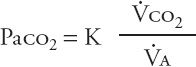
In this equation, ![]() co2 represents carbon dioxide production;
co2 represents carbon dioxide production; ![]() A, alveolar ventilation; and K, a constant. An increase in
A, alveolar ventilation; and K, a constant. An increase in ![]() co2 secondary to elevated metabolic activity is almost never the primary cause of hypercapnia because the respiratory system usually can compensate for the higher ventilatory requirement. When other elements of the respiratory system impair alveolar ventilation, however, increases in
co2 secondary to elevated metabolic activity is almost never the primary cause of hypercapnia because the respiratory system usually can compensate for the higher ventilatory requirement. When other elements of the respiratory system impair alveolar ventilation, however, increases in ![]() co2 (e.g., secondary to fever or sepsis) can contribute to hypercapnia. The central cause of hypercapnic respiratory failure remains inadequate alveolar ventilation.
co2 (e.g., secondary to fever or sepsis) can contribute to hypercapnia. The central cause of hypercapnic respiratory failure remains inadequate alveolar ventilation.
Alveolar ventilation is a physiologic process described by the equation:
![]()
![]() E (the volume of gas expired per minute, or “minute ventilation”) is a measurable quantity. The equation divides minute ventilation into two separate components: (1) alveolar ventilation (
E (the volume of gas expired per minute, or “minute ventilation”) is a measurable quantity. The equation divides minute ventilation into two separate components: (1) alveolar ventilation (![]() A), which participates in gas exchange, and (2) dead space ventilation (
A), which participates in gas exchange, and (2) dead space ventilation (![]() D), which does not.
D), which does not. ![]() D also may be viewed as “wasted” ventilation, that is, ventilation that does not reach the gas-exchanging areas of the lung. Rearranging this equation,
D also may be viewed as “wasted” ventilation, that is, ventilation that does not reach the gas-exchanging areas of the lung. Rearranging this equation,
![]()
This way of considering alveolar ventilation makes it clear that hypercapnia may occur via two distinct mechanisms: (1) a reduction in minute ventilation itself (absolute hypoventilation), or (2) an increase in dead space ventilation (relative hypoventilation).
Although mixed forms of hypoventilation occur, the distinction between absolute and relative hypoventilation is useful in separating patients with hypercapnic respiratory failure into two major categories: those with normal lungs and those with intrinsic disease of the lungs. Patients with normal lungs manifest hypercapnia because of inadequate minute ventilation caused by abnormalities in respiratory control (induced by disease or drugs), neuromuscular disorders involving the respiratory nerves and muscles, or chest wall abnormalities. Lung function may be normal in this group. Patients with abnormal lungs manifest hypercapnia because of the increased dead space (wasted) ventilation associated with maldistribution of ventilation and perfusion. The net result of these derangements is inadequate carbon dioxide clearance, even though minute ventilation (and respiratory drive) is normal or increased.
Combined forms of hypercapnic respiratory failure occur occasionally, such as in a patient with chronic obstructive pulmonary disease (COPD) who receives sedatives or narcotics (e.g., for anxiety or sleeplessness, or because of a misdiagnosis of left ventricular failure). Similarly, excessive diuretic use or other circumstances may cause hypokalemia, hypomagnesemia, or hypocalcemia and impair diaphragmatic contractility. Somewhat controversial is the relationship of excessive amounts of supplemental oxygen to absolute and relative hypoventilation and acute deterioration of patients with chronic hypercapnia (discussed below).
Regardless of the pathogenesis, the consequences of hypercapnic respiratory failure are the same. All patients with acute hypercapnia have hypoxemia, acidosis, an increase in pulmonary vascular resistance, and dilatation of the cerebral vessels. Arterial hypoxemia is an inevitable consequence of hypercapnia because, as alveolar Pco2 (PACO2) rises, alveolar Po2 (PAO2) and therefore arterial Po2 (PAO2) must fall. The alveolar–arterial gradient (“A–a gradient”) is a useful indicator of how much of a patient’s hypoxemia is attributable to hypoventilation itself. The A–a gradient (P(Aa)o2) is calculated as

At sea level, this equation becomes
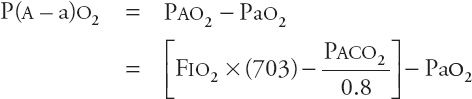
where PAO2 is the alveolar oxygen pressure, Pao2 is the arterial oxygen pressure, FIO2 is the fraction of oxygen in inspired air, PAH2O is the alveolar water vapor pressure (47 torr), and PACO2 is the alveolar CO2 pressure (which is equal to the PACO2). If hypercapnia alone is responsible for hypoxemia, the P(A–a)o2 is not widened. If this difference is widened, hypoxemia is likely due to cardiopulmonary disease. For example, in acute respiratory failure induced purely by drug overdose, the P(A–a)o2 might be normal, meaning that the hypoxemia might be fully explained by hypercapnia. However, if the patient has aspirated, zones of acute lung injury may create low ventilation–perfusion (![]() ) units leading to hypoxemia and an increased P(A–a)o2.
) units leading to hypoxemia and an increased P(A–a)o2.
ACIDOSIS
Acidosis is a direct consequence of hypercapnia, although patients with acute respiratory failure may have other reasons for acidosis. The severity of acidosis attributable to hypercapnia itself can be calculated using the equilibrium expression: CO
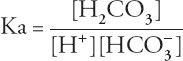
Rearranging the terms yields the Henderson-Hasselbalch equation:
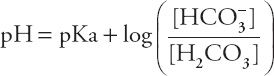
or
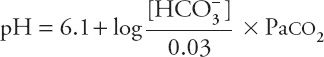
The HCO3– concentration in this equation is the actual concentration in the blood, not the concentration reported in the typical chemistry panel, which is measured after all the CO2 has left the serum. If the actual HCO3– concentration determined by the Henderson-Hasselbalch equation were normal, one could conclude that metabolic disorders are not contributing to the acid–base disorder. Although this might seem like a daunting mathematical task to perform at that bedside, a happy coincidence makes it quite easy. As it turns out, if the actual HCO3– concentration is normal, and the pH is between 7.0 and 7.5, the complicated Henderson-Hasselbalch equation comes very close to a straight line, with the formula:
![]()
Within these limits, an acute change in Paco2 of 10 torr changes (in the opposite direction) the blood pH by 0.08. Changes in pH not predicted by this equation must be attributed to causes other than acute hypercapnia.
Regardless of the cause, alveolar hypercapnia, hypoxemia, and arteriovenous acidosis all contribute to constriction of pulmonary resistance vessels and an increased pulmonary arterial pressure. This can lead to a higher work requirement for the right ventricle and right ventricular failure. The same factors cause dilatation of cerebral resistance vessels and increases in intracranial pressure. For this reason, hypercapnic respiratory failure may manifest itself by symptoms such as disorientation, personality changes, coma, headache, papilledema, and asterixis.
MANAGEMENT
The primary goals of management in acute hypercapnic respiratory failure are to (1) prevent respiratory arrest in patients who are rapidly decompensating; (2) restore adequate gas exchange; and (3) treat the disorder(s) responsible for inducing respiratory failure. These goals can be pursued simultaneously.
Supplemental Oxygen
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


