37 Inheritable Cancer Syndromes
Evelien Dekker, Frank G.J. Kallenberg, Joep E.G. IJspeert, and Barbara A.J. Bastiaansen
37.1 Introduction
Colorectal cancer (CRC) results from genetic factors as well as environmental influences and their interaction. Most CRC cases are so called “sporadic” and seem mainly caused by interplay of environmental influences such as diet, smoking, and lifestyle factors. In a small proportion of patients, the CRC is caused by a genetic predisposition.
A family history of CRC is common among the general population. In the Western world, approximately 5 to 10% of adults have a first-degree relative with colorectal cancer, resulting in an increased risk for CRC depending on the number and age of affected relatives. 1 Up to one-third of patients with CRC appear to have increased familial risk, likely related to inheritance (“familial CRC”), but only approximately 5% of all colorectal cancers have a clear, well-defined, inherited genetic predisposition.
Inheritable cancer syndromes can be subdivided in nonpolyposis and polyposis syndromes (▶Fig. 37.1). Lynch syndrome is a nonpolyposis syndrome caused by a dysfunction of the DNA mismatch repair system as a result of a germline mutation. Multiple adenomatous polyps may be caused by familial adenomatous polyposis (FAP), with a classical type (classical FAP) and a les profound type (attenuated FAP [AFAP]), and MUTYH-associated polyposis (MAP). Serrated polyposis syndrome (SPS) is a clinical diagnosis in patients with many serrated polyps, but the genetics are not yet known. Other, rare types of inheritable hamartomatous polyposis syndromes are Peutz–Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS), and Cowden’s syndrome (CS).
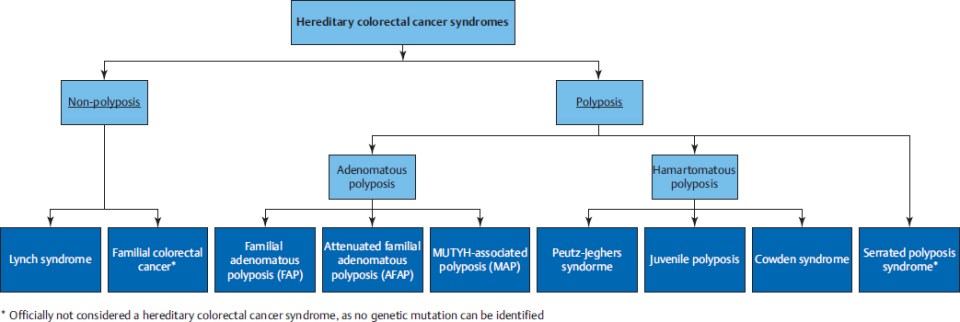
Diagnosing an inheritable cancer syndrome is important for several reasons: to provide an optimal surveillance strategy to prevent CRC, to provide optimal surveillance for extracolonic cancers if applicable, to provide optimal treatment in case of incident CRC, and to provide appropriate advice for relatives at risk to prevent CRC.
In general, polyposis syndromes are easily diagnosed as the number of polyps alerts the physician to think of a genetic syndrome, and the type of polyps might lead directly to the diagnosis. Lynch’s syndrome, however, is easily missed as those patients have few adenomas and those adenomas morphologically resemble sporadic lesions. Therefore, besides a family history for CRC and Lynch-associated cancers as well as the age of the patient having cancer as hallmarks for Lynch’s syndrome, systematic molecular analysis of tumor tissue is nowadays used to improve the diagnosis of this genetic syndrome.
Once diagnosed, each syndrome has its specific risks and appropriate surveillance strategy in an effort to prevent CRC and extracolonic cancers, also discussed in this chapter (▶Table 37.1).
37.2 Nonpolyposis Syndromes
37.2.1 Lynch’s syndrome
Genetics
Lynch’s syndrome is the most common inherited CRC syndrome and accounts for approximately 3% of newly diagnosed cases of CRC and 2% of endometrial cancer. It is an autosomal dominant disorder that is caused by a germline mutation in one of several DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2) or loss of expression of MSH2 due to a deletion in the EPCAM gene. The term hereditary nonpolyposis colorectal cancer (HNP-CC) refers to patients and/or families who fulfill the Amsterdam criteria for Lynch’s syndrome, and stems from the era when the genetic cause for this condition was unknown.
The role of the DNA MMR system is to maintain genomic integrity by correcting errors in base pairing during DNA replication. Inactivation of both alleles of one of the MMR genes leads to defective MMR and microsatellite instability (MSI). Patients with Lynch’s syndrome and CRC have a germline mutation in one allele of a MMR gene; the second allele is inactivated by mutation, loss of heterozygosity, or epigenetic silencing by promoter hypermethylation. Biallelic inactivation of MMR genes in a cell results in defective repair of DNA mismatches occurring during replication, causing genomic instability. DNA mismatches most commonly occur in regions of repetitive nucleotide sequences, which are called microsatellites. Thus, a characteristic of loss of mismatch repair in cancers is the expansion or contraction of these microsatellite regions in the tumor compared with normal tissue. This is called MSI, and characteristic of Lynch-associated cancers.
However, MSI is not specific for Lynch’s syndrome, and approximately 15% of sporadic CRCs also demonstrate MSI. Sporadic MSI-high cancers develop through somatic promoter methylation of MLH1, leading to loss of MLH-1 function and frequently carry mutations in the BRAF gene.
Characteristics
Individuals with Lynch’s syndrome are at an increased risk of CRC, endometrial cancer, and several other malignancies.
The lifetime risk of CRC in Lynch’s syndrome is between 25 and 70%, varies by genotype, and may be influenced by ascertainment bias. 2 , 3 It is diagnosed at a considerably younger age than sporadic cancer, with an average 44 to 61 years versus 69 years. 4 Besides, individuals with Lynch’s syndrome are at increased risk for synchronous and metachronous CRCs.
Most CRCs in Lynch’s syndrome are located in the right colon. It develops from adenomas, which have no specific endoscopic hallmarks to be discriminated from sporadic adenomas. The most important feature to be considered for adequate preventive measures is their behavior: in Lynch’s syndrome, the adenoma–carcinoma sequence is accelerated and estimated at 3 years, as opposed to 10 to 15 years for sporadic lesions. 5 CRCs in patients with Lynch’s syndrome show a diminished response of 5-fluorouracil–based chemotherapy, but interestingly, the overall 5-year survival from CRC in Lynch’s syndrome is higher when compared with sporadic CRC.
The most common extracolonic tumor in Lynch’s syndrome is endometrial cancer. The risk of endometrial cancer varies depending on the MMR mutation, and is highest for MSH-2 mutation carriers. 3 Individuals with Lynch’s syndrome are also at increased risk of cancer of the small bowel, stomach, ovaries, transitional cell cancer of the renal pelvis and ureter, hepatobiliary system, brain (glioma), and sebaceous neoplasms.
Diagnosis
Lynch’s syndrome should be suspected in patients with synchronous or metachronous CRC, CRC before the age of 50, multiple Lynch’s syndrome–associated cancers and in families showing clustering of Lynch’s syndrome–associated cancers. A pathogenic germline mutation in the MMR or EPCAM gene makes a definitive diagnosis of Lynch’s syndrome.
However, Lynch’s syndrome is largely underdiagnosed. Traditionally, a family history of colorectal and other Lynch-associated cancers was the primary tool to identify Lynch’s syndrome (Amsterdam criteria and Bethesda criteria, ▶Table 37.2). Nowadays, tumor testing for MSI and immunohistochemistry for loss of MMR proteins can be used as an additional tool for identification of Lynch’s syndrome. To optimize detection of Lynch’s syndrome, prediction models (e.g., PREMM) or systematic tumor testing for all newly diagnosed CRCs or all CRCs under age 70 are advocated. 6
Screening, Surveillance, and Treatment
Individuals with Lynch’s syndrome are advised to undergo CRC surveillance colonoscopies every 1 to 2 years beginning at age 20 to 25, or 5 years prior to the earliest age of CRC diagnosis in the family, whichever comes first. The main reason for frequent endoscopies is the accelerated adenoma–carcinoma pathway, and frequent colonoscopy screening, one or two yearly, was associated with a lower risk of CRC than colonoscopy screening every 2 to 3 years. 7 Quality of the colonoscopy is of utmost importance, ensuring optimal detection and complete resection of all precursor lesions. Upon detection of a CRC in a Lynch patient, a more radical surgical resection (e.g., subtotal colectomy in case of a right-sided carcinoma) should be discussed to prevent a metachronous CRC.
Women with Lynch’s syndrome are also advised regular surveillance for endometrial and ovarian cancer by a gynecologist. Optimal surveillance strategies are debated and evidence is very limited, but generally include pelvic examinations, cancer antigen 125 (CA-125) testing, transvaginal ultrasound, and endometrial biopsy. The advised frequency is yearly, starting between the age of 25 and 40 years. As surveillance strategies for endometrial carcinoma are unreliable, prophylactic hysterectomy with or without ovariectomy should be discussed with female Lynch’s patients after childbearing age.
Data on the effectiveness of surveillance programs for other Lynch-associated cancers are lacking. Surveillance schemes may include upper endoscopy and treatment of Helicobacter pylori infection when detected (once or regularly), urinalysis for tumor cells, and careful skin examination.
37.2.2 Familial CRC
Whereas one in four patients with CRC have a family history for this type of cancer, only approximately 4% are diagnosed with an inheritable cancer syndrome (▶Fig. 37.2). Individuals from families not diagnosed with an inheritable syndrome are at increased risk of developing CRC, but not as high as with the inheritable syndromes. Having a single affected first-degree relative (i.e., parent, child, sibling) increases the risk of developing CRC approximately twofold over that of the general population. 8 , 9 , 10 The risk is further increased if two first-degree relatives have CRC or if the index case is diagnosed before the age of 50, and those families have a clinical diagnosis of “familial CRC.”

As the adenoma–carcinoma sequence is not accelerated in these patients, surveillance colonoscopies are advised with 5-yearly intervals. Starting age is debated in literature, but generally 45 years with 5-yearly intervals.
37.3 Polyposis Syndromes
37.3.1 Familial Adenomatous Polyposis
Genetics
Familial adenomatous polyposis (FAP) has an incidence of 1:6,850 to 1:23,700 live births. 11 , 12 , 13 It is an autosomal dominantly inherited disease, caused by a germline mutation in the adenomatous polyposis coli (APC) gene, located on chromosome 5q21–q22. 14 In most cases, FAP is inherited from one of both parents, but in 25% cases it is caused by a “de novo” mutation. 11 In 20% of these “de novo” patients the APC mutation is found in only a part of all body cells, which is called mosaicism. 15 , 16 Furthermore, in approximately 20% of patients with a clinical diagnosis of FAP, no APC mutation can be identified. 17 , 18
Characteristics
FAP is characterized by the development of hundreds to thousands colorectal adenomas starting from puberty, which, without preventive measures, in nearly 100% will progress to CRC at the average age of 39 years (▶Fig. 37.3a, b). 19 This high cancer risk is not caused by an accelerated adenoma–carcinoma sequence (such as in Lynch’s syndrome), but is due to the high number of colorectal adenomas.

In many of these patients, extracolonic manifestations are also present. Of these, duodenal adenomas are most frequently encountered with a lifetime prevalence up to 90%. 20 These adenomas can be found throughout the duodenum and even in the proximal jejunum, but these usually appear at the ampulla of Vater or the periampullary region (▶Fig. 37.3c). 21 Likewise, this results in an increased lifetime risk of both duodenal and ampullary cancer of 3 to 10% at an average age of 44 years. 22 , 23 , 24 Another frequent finding in the upper gastrointestinal (GI) tract is fundic gland polyps, located in the fundus or body of the stomach (▶Fig. 37.3d). 25 These are small and sessile dilated glands, which rarely progress to cancer. Gastric adenomas are less common than fundic gland polyps, are typically located in the antrum, and have a low risk of progression to cancer. The most severe extraintestinal manifestations are desmoid tumors, occurring in approximately 15% of patients with FAP. These lesions are found in the skin or abdomen, where they can obstruct or perforate organs. Today, this is the leading cause of death in FAP patients together with duodenal cancer. 26 Other, but less frequent, extracolonic manifestations associated with FAP are, among others, sebaceous or epidermoid cysts, lipomas, osteomas, fibromas, supernumerary teeth, juvenile nasopharyngeal angiofibromas, adrenal adenomas, and congenital hypertrophy of the retinal pigment epithelium.
Related posts:
 35 Colorectal Polyps and Cancer Screening/Prevention
35 Colorectal Polyps and Cancer Screening/Prevention
 39 Lower Intestinal Bleeding Disorders
39 Lower Intestinal Bleeding Disorders
 40 Anorectal Diseases
40 Anorectal Diseases
 38 Inflammatory Bowel Disease and Microscopic Colitis
38 Inflammatory Bowel Disease and Microscopic Colitis
 36 Advanced Colorectal Polyps and Early Cancer Resection
36 Advanced Colorectal Polyps and Early Cancer Resection
 21 Hybrid, Natural Orifice, and Laparoscopy-Assisted Endoscopy: New Paradigms in Minimally Invasive Therapy
21 Hybrid, Natural Orifice, and Laparoscopy-Assisted Endoscopy: New Paradigms in Minimally Invasive Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


