Fig. 67.1
Ultrasonography (a) and colour Doppler (b) showing cavernomatous transformation of the portal vein in a 14-year-old boy with portal vein thrombosis. Liver MRI illustrating coronal (c), (d) and axial (e) T 2-weighted HASTE images in the same patient. HASTE half-Fourier single-shot turbo spin echo (Courtesy of Prof. Dr. Voet, Department of Ultrasonography, and Dr. N. Herregods, Department of Radiology, Ghent University Hospital, Belgium)
Management
Anticoagulation therapy can be considered for patients with a well-documented prothrombotic condition. In patients with idiopathic chronic EHPVO, there is no role for anticoagulant therapy. Insufficient evidence exists in favour of interventional therapy such as local thrombolysis [32].
For the management of portal hypertension caused by EHPVO refer to Chap. 68 “Portal Hypertension in Children”.
Hepatic Artery Anomalies
Ischaemic Cholangiopathy
Ischaemic cholangiopathy has been defined as focal or extensive damage to bile ducts due to impaired blood supply [37] . Unlike the hepatic parenchyma which has a dual blood supply from the hepatic artery and PV, the biliary system depends only on the arterial blood supply [3, 37]. Ischaemic bile duct injury may occur when small hepatic arteries or the peribiliary plexus are injured, or when all possible arterial blood supply is interrupted as in the case of hepatic artery thrombosis after liver transplantation [37]. Conditions associated with ischaemic cholangiopathy are iatrogenic factors (hepatic arterial chemotherapy [38, 39], abdominal radiation [40], liver transplantation [41]) and systemic diseases (panarteritis nodosa [42, 43], paroxysmal nocturnal haemoglobinuria [44]). In the acute stage, patients can present with pain, fever, and jaundice, with or without bacterial cholangitis. In the further course, localized or diffuse bile duct stenosis can occur with variable presentation going from no clinical signs to progressive or fluctuating jaundice, itching, fatigue, or bacterial cholangitis and eventual development of portal hypertension [37].
Pseudoaneurysm of the Hepatic Artery
Hepatic artery pseudoaneurysm (HAP) accounts for 12–20 % of all visceral aneurysms [45, 46]. HAPs can be found incidentally, but rupture of the aneurysm can be the first clinical manifestation with abdominal pain, gastrointestinal haemorrhage, or haemobilia [47]. Although HAPs can resolve spontaneously by thrombosis, the reported risk of rupture ranges from 14 to 80 % [45, 48]. Hence, once the diagnosis is confirmed, the aneurysm should be treated regardless of the symptoms. This is done by direct percutaneous injection with thrombin or glue, transarterial embolization or stent placement, or surgical approach [45].
Abnormalities of the Sinusoidal Blood Flow
Pericellular Fibrosis
The hepatic sinusoids comprise one of the largest-calibre vascular beds in the body. Impairment of blood flow through this vascular bed results in a major loss of physiologic function, with profound influence on homeostasis for the entire human organism [49]. The most common cause of sinusoidal blood flow obstruction is cirrhosis leading to a decrease in sinusoidal fenestrations, deposition of subendothelial basement membrane and collagen, loss of hepatocellular microvilli, increased expression of endothelial factor VIII and binding of ulex europaeus agglutinin (UEA)-1 lectin to endothelial cells [50, 51]. Pericellular fibrosis is commonly seen in alcoholic liver disease, chronic passive congestion, nonalcoholic fatty liver disease, Gaucher’s disease, congenital syphilis, and vitamin A toxicity [3].
Physical Occlusion of the Sinusoids
In sickle cell disease, sinusoids can become packed with sickled red cells and erythrophagocytes leading to parenchymal necrosis [52]. In disseminated intravascular coagulation and eclampsia, fibrin deposits may occlude the sinusoids. When these lesions are severe, widespread infarction might occur. Furthermore, the sinusoids might become infiltrated by mast cells in mastocytosis, Gaucher’s cells, metastatic tumour cells, and leukaemia or lymphoma cells [3].
Peliosis Hepatis
Peliosis hepatis is a rare condition in which the sinusoidal dilatation is primary [53]. The liver contains blood-filled cystic spaces, either non-lined or lined with sinusoidal endothelial cells [54]. The pathogenesis of peliosis hepatis is unknown [49]. In adults, peliosis hepatis can be induced by anabolic steroids, azathioprine, oral contraceptives, 6-thioguanine and 6-mercaptopurine, or presents in the context of chronic underlying disorders such as malnutrition [55], leukaemia [56], tuberculosis [57], vasculitis [58], cystic fibrosis , or human immunodeficiency virus (HIV) infection [59]. Peliotic lesions found in acquired immunodeficiency syndrome (AIDS) and other immunosuppressed patients are caused by bacterial organisms (Bartonella species) [56, 60]. In children, peliosis hepatis also occurs most frequently in association with chronic underlying conditions such as cystic fibrosis [61], malnutrition [62], Fanconi anaemia [63], adrenal tumours [64], Marfan syndrome [65], congenital cardiopathy [66], myotubular myopathy [67], or renal transplantation [68]. Four pediatric cases have been published without underlying systemic disorder. In these cases, there seemed to be an association with Escherichia coli infection suggesting a direct role of E. coli toxins in causing endothelial damage [69–71]. The definitive diagnosis of peliosis hepatis is based on histological findings but should be suspected when ultrasonography reveals hypoechogenic areas involving the whole liver in association with intraperitoneal fluid and normal Doppler signals [70].
Hepatic Vein Anomalies
Budd–Chiari Syndrome (BCS)
BCS is defined as hepatic venous outflow obstruction at any level from the small hepatic veins to the junction of the inferior vena cava and the right atrium, regardless of the cause of obstruction. Outflow obstruction caused by hepatic veno-occlusive disease and cardiac disorders is excluded from this definition. BCS can be classified as primary due to an endoluminal venous lesion (thrombosis or webs) or secondary due to intraluminal invasion by a parasite or malignant tumour or extraluminal compression by an abscess, cyst, or solid tumour [72] . In primary BCS, an underlying prothrombotic disorder or established risk factor for venous thrombosis is often present. In adults, myeloproliferative diseases account for half of the cases of BCS [73, 74]. Table 67.1 gives an overview of predisposing conditions for BCS. The role of hyperhomocysteinaemia and primary protein C, protein S, or antithrombin deficiency is unclear because liver disease obscures recognition of these disorders [75]. BCS is uncommon with an estimated incidence of 1 in 2.5 million persons per year [76]. It is very rare in children and has a wide variety of predisposing causes. Young children (< 10 years of age) account for 1–7 % of all cases of BCS [77], whereas in endemic areas, children younger than 10 years may compose 16 % of the total cases [78]. Hepatic venous outflow tract obstruction leads to increased sinusoidal pressure, sinusoidal congestion, hepatomegaly, hepatic pain, portal hypertension, and ascites . Elevated sinusoidal pressure leads to perisinusoidal necrosis of hepatocytes in the centrilobular region and eventually to irreversible liver damage, cirrhosis, or liver failure [79].
Table 67.1
Predisposing conditions for Budd–Chiari syndrome
Inherited conditions |
Factor V Leiden mutation |
G20210A prothrombin gene mutations |
Hyperhomocysteinaemia |
Primary protein C or protein S deficiency |
Antithrombin deficiency |
Acquired conditions |
Myeloproliferative disorders (V617 JAK2 positive) |
Antiphospholipid syndrome |
Behcet’s disease |
Paroxysmal nocturnal haemoglobinuria |
Environmental factors |
Oral contraceptive use |
Toxins like heavy metals, aflatoxins, etc. |
A diagnosis of BCS should be considered in any patient who presents with acute or chronic liver disease as the clinical manifestations can be extremely diverse. The majority of patients present with the typical triad described by George Budd in 1845: right upper quadrant pain, hepatomegaly, and ascites. Oedema of the lower extremities is also a common finding [72] . Asymptomatic BCS accounts for 15–20 % of cases [80]. Jaundice, gastrointestinal bleeding, and hepatic encephalopathy are less common [75]. The diagnosis of BCS is established upon demonstration of obstruction of the hepatic venous outflow tract. US combined with Doppler imaging has a diagnostic sensitivity of more than 75 % and should be the first line of investigation [81, 82]. Hepatic veins devoid of flow signal, collateral hepatic venous circulation, a spider web appearance usually located in the vicinity of the hepatic vein ostia and stagnant, reversed, or turbulent flow can all be indicative of BCS [83, 84]. When adequate ultrasonography is technically difficult or when diagnostic features cannot be demonstrated, CT or MRI should be performed. The latter is preferred in children due to absence of radiation exposure. Only in a minority of cases, retrograde cannulation of the hepatic veins for X-ray venography will be ultimately necessary for diagnosis. This technique allows the assessment of the extent of venous outflow obstruction and allows for pressure measurements (Fig. 67.2). Concurrent liver biopsy can contribute in confirming the diagnosis and ruling out other causes such as veno-occlusive disease and cirrhosis of other etiologies [85]. Once the diagnosis of BCS is established, the patient should be investigated for underlying prothrombotic conditions and a haematologic work up for myeloproliferative disorder should be performed. The rarity of BCS in children often means that the disease is diagnosed in its later stages by which time irreversible pathology may be present [77] .
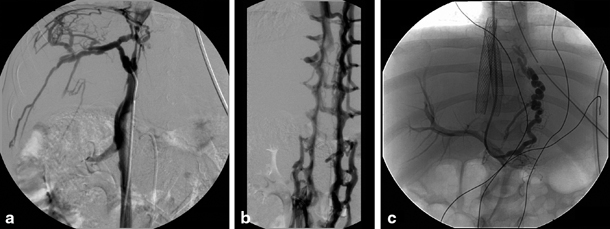
Fig. 67.2
Congenital stenosis of the inferior caval vein with Budd–Chiari syndrome in a 4-year-old child. a Cranial cavography with catheter tip at caudal site of inferior caval vein stenosis with visualization of collateral circulation at the level of the liver capsule. b Caudal cavography illustrating collateral circulation via the lumbar veins caused by the more cranially positioned stenosis. c Placement of stent in inferior caval vein and TIPSS with direct venography of oesophageal varices. TIPSS transjugular intrahepatic portosystemic stent shunting. (Courtesy of Dr. P. Van Langenhove and Prof. Defreyne, Department of Interventional Radiology, Ghent University Hospital, Belgium)
The management of BCS in adults consists of anticoagulation, thrombolytic therapy, and angioplasty with or without stenting, transjugular intrahepatic portosystemic shunts (TIPSSs), and surgically fashioned portosystemic shunts [77, 86]. These approaches have, to variable degrees, been extrapolated for use in pediatric BCS. Early referral has the best possible outcome by creating the opportunity of re-establishing native hepatic venous outflow tract without surgery. Thrombolytic therapy might be successful in dissolving fresh thrombi and can be combined by transjugular balloon angioplasty. Local infusion of thrombolytic therapy in a partially recanalized vein with appreciable flow in a patient who presents earlier than 4 weeks has been associated with a consistently successful outcome in adult patients [87]. Maintenance of flow can be ensured by long-term anticoagulation and treatment of the underlying haematologic disorder if one is identified. TIPSSs have been successfully used in children with favourable long-term results [88, 89]. Percutaneous stent placement has also played a role in the management of pediatric BCS [90, 91] (Fig. 67.2). In a late presentation with established hepatic cirrhosis and complications of portal hypertension, liver transplantation is the only option that can offer an excellent chance of long-term survival. Reports on the long-term outcomes for orthotopic liver transplantation (OLT) specifically for BCS in children are scarce and limited to a few sporadic case reports [92, 93] .
Veno-Occlusive Disease (VOD)
VOD , also known as sinusoidal obstruction syndrome, is a severe and potentially fatal liver disease originally described in Jamaican drinkers of pyrrolizidine alkaloid-containing bush tea. It is now seen predominantly, but not exclusively, in patients undergoing haematopoietic stem cell transplantation (HSCT) [94]. The incidence of VOD after HSCT varies between 10 and 60 % in different series [95]. VOD usually presents in the first 30 days after HSCT [96, 97]. The clinical course is characterized by rapid weight gain, jaundice, abdominal pain, hepatomegaly, and ascites . Encephalopathy may develop. The most commonly used diagnostic criteria for VOD are the (modified) Seattle criteria [97] and the Baltimore criteria [96] illustrated in Table 67.2. Doppler ultrasound can further support the diagnosis by showing evidence of decreased or reversed portal venous flow. The pathogenesis of VOD is most likely due to a primary injury to the endothelial cells of sinusoids and small venules by chemotherapy and radiation. Many of the cytotoxic agents used in HSCT are metabolized in the liver, including cyclophosphamide and busulfan. It is thought that depletion of glutathione in zone 3 hepatocytes and sinusoidal cells plays an important role in the initiation of the damage [98]. A number of markers of endothelial injury and adhesion molecules are upregulated in patients with VOD. These include plasma thrombomodulin, P- and E-selectins, tissue factor pathway inhibitor, soluble tissue factor and plasminogen activator inhibitor (PAI-1). Increased serum levels of PAI-1 is both a diagnostic and prognostic marker for VOD [99–102]. Histopathologically, the early lesion is subintimal oedema and haemorrhage involving the hepatic venules with fibrin deposits. This leads to sinusoidal congestion and dilatation with (mainly centrizonal) hepatocellular necrosis. In the further course, fibrous obliteration occurs of the venular walls with atrophy of perivenular hepatocytes and development of sinusoidal fibrosis potentially leading to venocentric cirrhosis [3, 103]. Risk factors for VOD include advanced malignancy and pre-existing liver disease which might be the consequence of previous hepatotoxic chemotherapy, previous abdominal irradiation, or iron overload due to repeated blood transfusions [96, 97, 104]. Busulfan, especially in combination with cyclophosphamide increases the risk of VOD [104, 105] . Targeted dosing of busulfan and pharmacokinetic monitoring of cyclophosphamide and its metabolites could be effective in reducing the risk of VOD [106]. The risk of VOD is higher in allogeneic compared to autologous transplants, with a higher incidence in unrelated and mismatched donors versus sibling donors [96, 97, 104]. High-level evidence from randomized controlled trials supporting prophylaxis for hepatic VOD is scarce [107]. Ursodeoxycholic acid might reduce the incidence of hepatic VOD, but trial results are conflicting and did not show any survival benefit [108–111]. The same holds true for trials on low-dose heparin infusion for VOD prophylaxis [112, 113]. Furthermore, trials on enoxaparin, glutamine, and fresh frozen plasma (FFP) all failed to demonstrate efficacy on reduction of VOD or overall mortality. Supportive treatment of VOD focusses on maintaining intravascular volume and renal perfusion without increasing extravascular fluid accumulation. Avoidance of exposure to hepatotoxic drugs, fluid and sodium restriction, and diuretics are important in the care of a patient with VOD. Again, high-level evidence is lacking on treatment options. There is substantial evidence for the efficacy of defibrotide, a polydisperse mixture of single-stranded oligonucleotide with antithrombotic and fibrinolytic effects on microvascular endothelium, in the treatment of VOD [114, 115]. Defibrotide can be used safely in pediatric patients [116]. Despite emerging therapies such as defibrotide, VOD remains a much feared transplant complication with unfavourable prognosis. Therefore, risk stratification of patients before HSCT is essential to minimize severe VOD and improve transplant outcome. Improved understanding of risk factors will enable to offer high-risk patients a reduced intensity conditioning regimen or T cell depletion to minimize their risk for transplant mortality. As PAI-1 is now recognized to be associated with the pathogenesis of VOD, serial monitoring of blood PAI-1 levels could offer a new diagnostic and potential prognostic tool for this disease [117] .
Seattle criteria (modified) [97] |
Presence of at least two of the following before day 20 after HSCT |
Bilirubin > 35 μmol/l |
Hepatomegaly or right upper quadrant pain |
Ascites or unexplained weight gain of 2 % above baseline |
Baltimore criteria [96] |
Bilirubin > 35 μmol/l in the first 20 days after HSCT and at least two of the following: |
Hepatomegaly |
Ascites |
Weight gain (> 5 % compared to pre-transplant) |
Congestive Cardiac Failure
Congestive cardiac failure is associated with dilatation of sinusoids and atrophy of perivenular hepatocytes which can lead to fibrosis and occasionally nodular regenerative hyperplasia (NRH) [118].
Hepatic Vascular Shunts
Arteriovenous Malformations
Arterioportal shunts may be congenital (in hereditary haemorrhagic telangiectasia) or acquired (blunt or penetrating trauma, percutaneous liver biopsy, cirrhosis) and consist of a communication between the hepatic artery and the portal venous system [3, 16, 21]. Most congenital arterioportal fistulas are symptomatic within the first year of life. Hepatofugal flow develops in the arterialized PV which can lead to portal hypertension, hypersplenism, varices, ascites, and hypertensive enteropathy resulting in malabsorption and diarrhea [21] .
Hepatic arteriosystemic shunts are the rarest form of intrahepatic shunts connecting the hepatic artery (or other systemic arteries) and the hepatic veins. These shunts may be congenital or associated with hereditary haemorrhagic telangiectasia, hepatocellular carcinoma, or large haemangiomas [16]. Hepatic arteriosystemic shunts are usually localized in one lobe of the liver [21]. Arteriovenous fistulas can not only present clinically in neonates with congestive heart failure, anaemia, hepatomegaly, and portal hypertension but can also manifest later in childhood in the clinical setting of hereditary haemorrhagic telangiectasia with congestive heart failure, hepatic ischaemia, and portal hypertension [21].
Portosystemic Shunts
The complicated development of the inferior caval vein and the close relationship of its development with that of the vitelline veins may explain the occurrence of congenital portosystemic anastomoses. Portosystemic venous shunting causes elevated levels of galactose, bile acids, ammonia, and other nitrogenous substances in the plasma, some of which can affect the brain [16, 119]. The age of onset of encephalopathy is variable but related in part to the volume and duration of the shunt and the presence of concomitant liver disease [119]. Both extrahepatic and intrahepatic portosystemic shunts have been described [21, 120].
Extrahepatic portosystemic venous shunts are also known as Abernethy malformations described in 1793 [1, 121–124]. Morgan and Superina classified extrahepatic portosystemic shunts into two types [121, 124]. In type I, there is a complete diversion of the portal blood flow in the caval vein, with congenital absence of the PV. Type Ia has absent PV, with the SV and SMV entering in systemic veins separately. These patients are usually girls with cardiac or other congenital anomalies including biliary atresia, oculo-auriculovertebral dysplasia, situs inversus, and polysplenia. Hepatic masses are frequent, usually described as focal nodular hyperplasia (FNH) , but sometimes hepatoblastoma , hepatocellular carcinoma, and adenoma. In type Ib, the SV and SPV join into a PV which then drains into a systemic vein. This form usually occurs in boys without the associated anomalies or hepatic masses [1, 3]. In type II, the PV is intact, but some of the portal flow is diverted into the caval vein through a side-to-side extrahepatic communication, which is congenital, usually isolated, and mostly seen in boys (type IIa) or acquired, most often induced by portal hypertension (type IIb) [21].
Congenital intrahepatic portosystemic shunts are abnormal intrahepatic connections between branches of the PV and the hepatic veins [3, 125] (Fig. 67.3). Intrahepatic portosystemic shunts which are acquired are most often associated with hepatic trauma or portal hypertension.
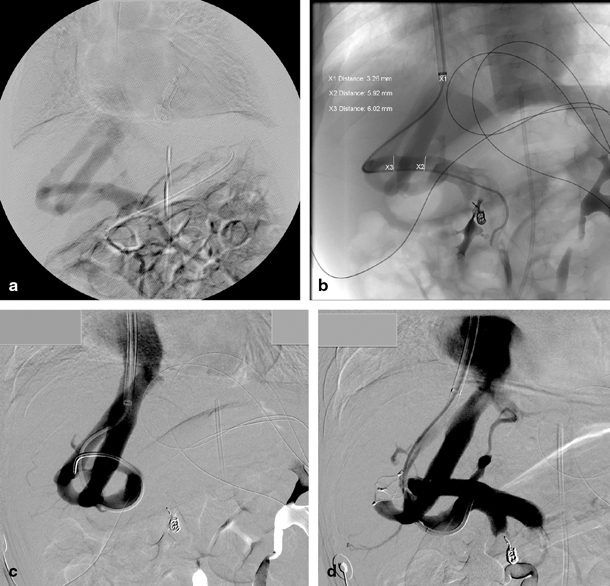
Fig. 67.3
A 5-month-old child with multiple intrahepatic portosystemic shunts. a Indirect mesentericography with visualization of multiple intrahepatic portosystemic shunts. b Direct portography in the same patient with guiding into the right hepatic vein. The child previously presented with acute haematemesis caused by a bleed from a pseudoaneurysm of the gastroduodenal artery which was coiled in a previous procedure. c Illustration of interconnections in the complex portosystemic shunt. d Direct portography after occlusion of one connection of the portosystemic shunt with vascular plug. (Courtesy of Dr. P. Van Langenhove and Prof. Defreyne, Department of Interventional Radiology, Ghent University Hospital, Belgium)
Hereditary Haemorrhagic Telangiectasia
Hereditary haemorrhagic telangiectasia (HHT) or Rendu–Osler–Weber disease is an autosomal dominant vascular disorder with variable penetrance with an estimated prevalence around 1 in 5000–8000 [126]. HHT is characterized by mucocutaneous telangiectases, recurrent epistaxis, and visceral arteriovenous malformations . Hepatic vascular malformations have been reported in 47 % of pediatric patients with HHT ranging from small telangiectases to discrete arteriovenous malformations [127].
Parenchymal Response to Vascular Injury
Nodular Regenerative Hyperplasia (NRH)
NRH is the major cause of non-cirrhotic portal hypertension in the Western world. It is a benign condition characterized by diffuse transformation of the liver parenchyma into small regenerative nodules distributed evenly throughout the liver with minimal or no fibrosis in the perisinusoidal or periportal areas [118]. NRH results from abnormalities in the portal hepatic and occasionally small hepatic blood flow giving rise to ischaemic atrophy and a secondary adaptive hyperplastic reaction of hepatocytes in regions with favourable blood flow [128, 129]. NRH occurs predominantly in older patients and is rather uncommon in children [130]. In a large series of 716 pediatric liver tumours , NRH was demonstrated in 4.5 % of the cases [131]. NRH is seen in conditions affecting the hepatic blood flow including solid-organ transplantation, bone marrow transplantation, vasculitic conditions, and can be associated with underlying autoimmune, inflammatory, and neoplastic diseases or HIV [49]. Immunosuppressive medications such as azathioprine, 6-mercaptopurine, and 6-thioguanine may induce NRH by damaging endothelial cells of small hepatic veins [132–134]. NRH presenting with progressive portal hypertension was described in six children treated with 6-thioguanine as maintenance therapy for childhood acute lymphoblastic leukaemia [135]. Imaging findings are relatively poor in sensitivity and specificity for NRH. A diffusely heterogeneous hepatic parenchyma may be the only imaging abnormality. Regenerative nodules are usually not visible on ultrasound [129] . On CT , regenerative nodules remain isodense or hypodense in both arterial and portal venous phases, distinguishing NRH from FNH and adenomas [130]. The significance of MRI in the diagnosis of NRH is still controversial with only few reports in the literature. Lesions appear hyperintense on T 1-weighted images and iso- or hypointense on T 2-weighted images [136]. The gold standard for diagnosis is histopathology demonstrating regenerative nodules consisting of hypertrophied hepatocytes centrally surrounded by atrophic hepatocytes peripherally. There is no or minimal perisinusoidal or portal fibrosis on reticulin staining and compression of the central veins by the regenerating nodules may be seen [137, 138]. The management of NRH mainly relates to the prevention and treatment of complications of portal hypertension.
Focal Nodular Hyperplasia (FNH)
FNH is a localized hyperplastic lesion in response to locally augmented arterial blood flow. The diagnosis of FNH is rarely made in the pediatric population [139]. It most often affects females between the age of 30 and 50 years [140, 141]. Most cases are found incidentally on abdominal imaging. Large subcapsular lesions might lead to vague abdominal pain. Complications such as rupture and intratumoral haemorrhage are extremely rare. Most lesions remain stable with a likelihood to regress with age, and there is no malignant potential [139]. On imaging, FNH should be differentiated from liver cell adenoma [142] or hepatocellular carcinoma [143]. The typical ultrasound finding in FNH is a well-demarcated homogeneous hypo- or isoechoic lesion with a central scar which can be seen in less than 20 % of cases. On colour Doppler, there is a central arterial structure with a spoke-wheel pattern of radiating smaller aberrant vessels [144]. CT shows typically a well-circumscribed iso- or hypodense lesion with rapid homogeneous intense enhancement in the arterial phase and gradual enhancement of the central scar in the portal venous phase [145]. On MR, a homogeneous lesion is seen that is isointense or slightly hypointense on T 1– and isointense or slightly hyperintense on T 2– weighted images. During the arterial phase, the typical FNH lesion becomes homogeneously hyperintense apart from the central scar which often exhibits avid enhancement in the delayed phase [146] (Fig. 67.4). In the presence of typical radiological findings there is usually no indication for liver biopsy. When performed, histopathology (Fig. 67.5) shows a non-encapsulated nodule with a central stellate fibrous region containing large vessels from which there are radiating septa. The parenchyma between the septa exhibits essentially normal hepatocytes but with a thickened plate architecture characteristic of regeneration [140]. Patients with asymptomatic FNH are treated conservatively without need for further imaging if there is a typical radiological appearance of FNH. In case of symptoms, which are usually seen in large subcapsular lesions, or in case of atypical radiological features which do not allow to rule out malignancy, surgical resection can be indicated [139].
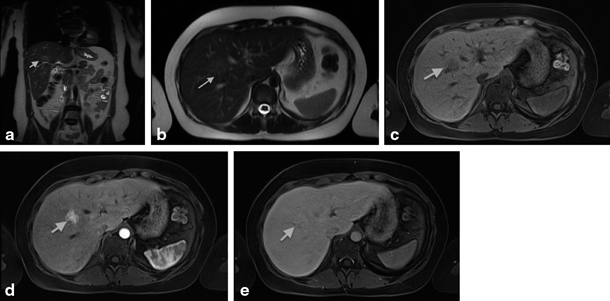
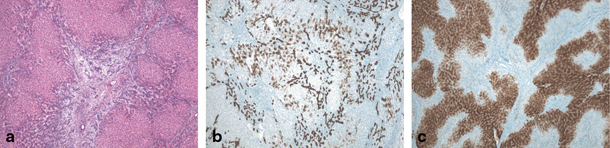
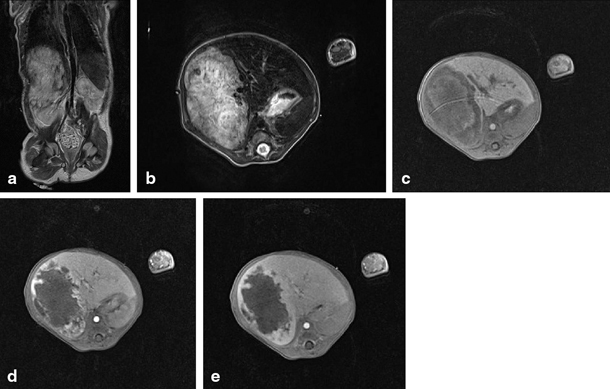
Fig. 67.4
MRI showing focal nodular hyperplasia (FNH) in a 16-year-old girl. This lesion is discretely hyperintense on T2 weighted images (a, b), hypointense on T1 weighted images (c), with intense early arterial phase enhancement (d) and isointense aspect compared to normal liver parenchyma in the portovenous phase (e). (Courtesy of Dr. N. Herregods, Department of Radiology, Ghent University Hospital, Belgium)
Fig. 67.5
Focal nodular hyperplasia (FNH) in 11-year-old girl. a H&E (40 ×) stained image showing fibrous scar and fibrous septa surrounding nodular hyperplastic parenchyma containing ductular reaction and multiple small arterial branches. b Cytokeratin 7 staining (40 ×) illustrating ductular reaction and cholate stasis in the lesion. c Typical ‘map-like’ pattern of FNH lesion on glutamine synthetase staining (40 ×). (Courtesy of Prof. L. Libbrecht, Department of Pathology, Ghent University Hospital, Belgium)
Benign and Malignant Vascular Tumors
Vascular tumours of the liver comprise a substantial portion (13 %) of all hepatic neoplasms in children [147]. Most of these lesions are benign. Infantile hepatic haemangioma (HH) is the most common benign tumour of the liver in infancy [148] . The terminology used in the literature is quite confusing and infantile HH is also called infantile haemangioendothelioma. Most of these haemangiomas remain asymptomatic and probably a substantial part remains undiagnosed [149]. An increasing number of hepatic haemangiomas is being identified on antenatal ultrasound [150]. Almost all patients with HH present before 6 months of age with most being diagnosed within the first 2 months [150–153]. The main symptoms are abdominal mass or distension. Other potential presentations include failure to thrive, high-output congestive heart failure, anaemia, thrombocytopenia (Kasabach–Merritt syndrome), respiratory distress, pulmonary hypertension, liver failure, and jaundice. In rare cases, spontaneous rupture has been described. Infantile HH express type 3 iodothyronine deiodinase that converts thyroid hormone to its inactive form. This can result in acquired hypothyroidism which is often seen in larger multifocal and diffuse HH and resolves with tumour involution [154–156]. Based on data from the Liver Haemangioma Registry, a division into three principal categories has been proposed: focal, multifocal, and diffuse lesions [149, 156]. Focal lesions could be considered as the hepatic variant of the cutaneous rapidly involuting congenital haemangioma which typically evolves during foetal life and is fully grown at birth. These lesions do not expand postnatally and are less commonly associated with accompanying cutaneous infantile haemangioma [152, 157, 158]. Because they develop antenatally, focal HH can be diagnosed prenatally [159–161]. Most focal HH are discovered as an abdominal mass in an otherwise healthy child [156]. Ultrasonography reveals a well-circumscribed mass with large feeding and draining vessels. On CT or gadolinium MRI, a well-defined, solitary, spherical tumour with centripetal enhancement and central sparing because of thrombosis, necrosis, or intralesional haemorrhage can be seen [149, 150, 162] (Fig. 67.6) . Multifocal and diffuse HH are considered as true infantile haemangiomas [156]. They are associated with cutaneous infantile haemangiomas in the majority of cases and characterized by the immunoexpression of glucose transporter (GLUT)-1 in liver tissue [156]. These lesions appear within the first weeks of life and are therefore not antenatally detected. The typical course in these lesions is one of rapid postnatal growth (0–12 months) followed by slow involution (1–5 years) [156]. Multiple well-defined spherical lesions are observed on CT, MRI, or ultrasound with intervening areas of normal hepatic parenchyma in multifocal HH, whereas the lesions in diffuse HH nearly totally replace the liver [156] (Fig. 67.7). Biopsy can be avoided in typical haemangioma (i.e. multifocal haemangiomas with cutaneous involvement or solitary haemangiomas, presenting in the first few months of life, with typical imaging findings) but should not be delayed in the face of diagnostic uncertainty or when children present with vascular tumours after infancy [150, 156]. Two histological subtypes have been described (Fig. 67.8). Type 1 HH are composed of capillary, sinusoidal and cavernous parts lined by plump endothelial cells with a bland cytological appearance [163]. Type 2 HH have areas composed of papillate tufting vascular channels that are lined by larger pleomorphic and hyperchromatic endothelial cells which exhibit more extensive cell proliferation and active mitosis [147]. The treatment of HH is controversial and the effects of the various forms of therapy are diverse and inconclusive [164]. Asymptomatic HH should be observed. Imaging studies of the brain and chest radiography are appropriate for patients with multifocal and diffuse HH [150] and thyroid function should be checked. In case of symptoms, medical treatment is indicated. Corticosteroids are most frequently used as pharmacological therapy. Early evidence suggests that propranolol, a nonselective β-blocker, may be as efficacious as corticosteroids in the treatment of infantile HH [165, 166] . Other pharmacological treatments which have also been reported to be effective are α-interferon therapy, chemotherapeutic agents, such as vincristine [167, 168], actinomycin D, and cyclophosphamide [169]. When there is no response to pharmacological treatment, arterial embolization, hepatic artery ligation, or surgical resection should be considered. Hepatic transplantation may be indicated in extremis when other treatment options are impossible or fail [156]. Infantile hepatic haemangiomas differ from cavernous haemangiomas which are usually asymptomatic in children. Being devoid of malignant potential, they are usually discovered as an incidental finding during abdominal imaging, most frequently between the fourth and the fifth decades of life. They are multiple in more than 50 % of cases and show a clear female predominance. Pathological examination reveals a focal tender mass formed by multiple vascular channels limited by a single layer of endothelial cells with thin fibrous stroma. In general, the blood circulation within these tumour vessels is slow. Morphologically, it is a well-defined lesion, possessing round or lobulated margins. There size usually remains stable and can vary from a few millimetres to more than 20 cm [143] (Fig. 67.9). Pediatric angiosarcoma, in contrast to HH, is a very rare but highly malignant tumour. It usually presents with a rapidly growing hepatic mass. The precise diagnosis may be difficult, even on a biopsy specimen [170]. Open biopsy of the tumour is therefore advisable. Most angiosarcomas develop after the first year of life. Chemotherapy and radiotherapy are notably inefficient in achieving tumour control, and the prognosis remains very poor. Radical resection or even liver transplantation should therefore be attempted if possible. Hepatic haemangioendothelioma is a rare neoplasm of endothelial origin having a clinical behaviour intermediate between haemangioma and angiosarcoma [171, 172]. Patients generally present from the second decade, and the tumour commonly affects liver, lung, skin, or bone but also other presentations are reported [173] .
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
