Fig. 68.1
Anatomy of the portal system
In this system, the central vessel is the portal vein, which is formed by the union of the splenic vein (SV) and the superior mesenteric vein (SMV), but receiving blood also from the inferior mesenteric vein (IMV), the gastric, and the cystic veins. The SMV is formed by tributaries from the small intestine, colon, and head of the pancreas, and irregularly from the stomach via the right gastroepiploic vein. The SVs originate at the splenic hilum and join near the tail of the pancreas with the short gastric vessels to form the main SV. The IMV carries blood from the left part of the colon and rectum and reaches the SV in its medial third. Anatomical variations include the IMV draining into the confluence of the SMV and the SV, and the IMV draining in the SMV (Fig. 68.1).
Immediately before reaching the liver, the portal vein divides into right and left main branches, and then ramifies further, forming smaller venous branches, and ultimately the portal venules. Each portal venule runs alongside a hepatic arteriole and the two vessels form the vascular components of the portal triad. These vessels ultimately merge into the hepatic sinusoids to supply blood to the liver. Three hepatic veins (right, middle, and left) drain the blood from the liver into the inferior vena cava (IVC) [10].
Pathophysiology of Portal Hypertension
The portal venous pressure is directly proportional to the portal blood flow and the hepatic resistance , according to Ohm’s law (∆P = Q × R, where ∆P is the variation of pressure along the vessel, Q is the blood flow, and R is the resistance to flow). Since portal vascular resistance is inversely proportional to the fourth power of the radius (Poiseuille’s equation), a small decrease in the vessel diameter produces a large increase in the portal vascular resistance and, in turn, in portal blood pressure. In the healthy liver, the intrahepatic resistance changes according to the variation of portal blood flow to keep portal pressure within normal limits. In fact, under physiological conditions, a rise in portal pressure is counteracted by sinusoidal dilatation, even in the presence of increased blood flow as can happen after meal ingestion [10, 11]. Most of the following statements made on PH come from experiments on animal models, such as the rat with a ligated portal vein or bile duct or with carbon tetrachloride-induced cirrhosis, and then confirmed in clinical studies carried out mainly in adults [11–13] .
Increase of Vascular Resistance
The portal venous system has a baseline portal pressure of 7–10 mmHg, and the hepatic venous pressure gradient (HVPG) ranges from 1 to 4 mmHg. PH is defined as a portal pressure greater than 10 mmHg or a gradient greater than 4 mmHg. In adults, a pressure gradient above 10 mmHg has been associated with esophageal varices (EV) formation, and with ascites and variceal bleeding if above 12 mmHg [14, 15].
The main pathogenetic factor in the development of PH is an increased vascular resistance (Fig. 68.2). Depending on the site in which it occurs, PH can be classified as extrahepatic (prehepatic and posthepatic) and intrahepatic. The latter may be further subdivided into three forms including presinusoidal (portal venules), sinusoidal (sinusoids), and postsinusoidal (terminal hepatic venules; central veins; Table 68.1).
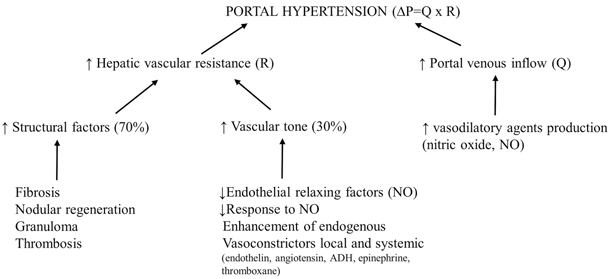
Fig. 68.2
Portal hypertension development according to Ohm’s law
Table 68.1
Classification and etiology of portal hypertension
Prehepatic |
Portal vein thrombosis |
Congenital stenosis or extrinsic compression of the portal vein |
Splenic vein thrombosis |
Artero-venous fistulae |
Intrahepatic presinusoidal |
Congenital hepatic fibrosis |
Chronic viral hepatitis (HBV and HCV) |
Primary biliary cirrhosis |
Myeloproliferative diseases (Hodgkin’s disease, leukemia) |
Focal nodular hyperplasia |
Idiopathic portal hypertension (IPH)/non-cirrhotic portal fibrosis (NCFP)/hepatoportal sclerosis |
Granulomatous diseases (schistosomiasis, sarcoidosis, tuberculosis) |
Amyloidosis |
Gaucher’s disease |
Polycystic liver disease |
Infiltration of liver hilum (independent of cause) |
Benign and malignant neoplasms |
Toxins and drugs (arsenic, vinyl chloride monomer poisoning, methotrexate, 6-mercaptopurine) |
Peliosis hepatis |
Rendu–Osler–Weber syndrome |
Chronic hepatitis |
Intrahepatic sinusoidal |
Liver cirrhosis (independent of cause) |
Wilson’s disease |
Hemochromatosis |
Storage diseases (fatty liver, glycogenosis type III, Niemann–Pick disease, α1-antitrypsin deficiency) |
Acute Hepatitis (viral and autoimmune) |
Hypervitaminosis A |
Intrahepatic postsinusoidal |
Veno-occlusive disease (VOD) |
Hepatic vein thrombosis (Budd–Chiari syndrome) |
Posthepatic |
Inferior vena cava obstruction (thrombosis, neoplasms) |
Right heart failure |
Constrictive pericarditis |
Tricuspid valve diseases |
In patients affected by chronic liver disease, though, increased vascular resistance is located at various intrahepatic levels.
The pathogenetic mechanism explaining the increased resistance in extrahepatic PH, where the blood flow is blocked by a mechanical obstruction, is quite obvious. Conversely, the pathogenesis is more complicated in intrahepatic PH, in which many factors, both mechanical and dynamic, may occur simultaneously [15].
Increase of Portal Blood Flow
The second factor contributing to the development of PH is an increase in blood flow, established through splanchnic arteriolar vasodilatation caused by an excessive release of endogenous vasodilators including nitric oxide (NO), glucagon, endothelin (activated by the vasoactive intestinal peptide), as well as by the activation of the sympathetic and the renin–angiotensin systems. These changes cause sodium and water retention, hypervolemia, renal hypoperfusion, and increase in cardiac output and splanchnic blood inflow, resulting in a hyperdinamic vascular status which characterizes the advanced stages of PH (Fig. 68.2).
Extra-hepatic Causes of PH
Prehepatic causes of increased resistance to flow include SV thrombosis, congenital atresia or stenosis of the portal vein, extrinsic compression (tumors), and portal vein thrombosis (PVT). In these disorders, the obstruction in the prehepatic portal venous system leads to an increased portal venous pressure [16].
The isolated obstruction of the SV (mainly due to thrombosis) usually results in left-sided PH (sparing the superior mesenteric district). In this rare clinical condition, the blood flows retrogradely through the short and posterior gastric veins and the gastroepiploic veins, leading to the formation of isolated gastric varices. The most common causes of SV occlusion are pancreatic diseases, such as pancreatic cancer, pancreatitis, or a pseudocyst. Although very rare in children, it should be considered in the presence of isolated gastric bleeding with normal liver function and unexplained splenomegaly. The diagnosis may be difficult, and splenectomy represents the treatment of choice in symptomatic patients [17–19].
PVT is the most prevalent cause of extrahepatic portal vein obstruction (EHPVO), and is the major cause of non-cirrhotic PH in children. Conversely, congenital abnormalities, such as portal vein stenosis, atresia, or agenesis, are relatively uncommon.
The etiology of PVT remains obscure in approximately 50 % of the cases whereas known etiologies include umbilical vein catheterization, omphalitis/umbilical sepsis, thrombophilia (acquired, hereditary), myeloproliferative disorders, surgery (splenectomy, liver transplantation), dehydration, and multiple exchange transfusions in the neonatal period [20–22].
In a multicenter Italian study including 187 pediatric patients diagnosed with EHPVO, it was shown that the condition is strictly associated with a neonatal disorder. The mean age at diagnosis was 4 years; 59 % were born preterm; 65 % had a history of umbilical catheterization; 82 % had associated illnesses, such as complications of prematurity (43.5 %), cardiac malformations (7.5 %), noncardiac malformations (8.5 %), deep infections (7 %), and hematological disorders (5.5 %). The patients were diagnosed upon detection of splenomegaly (39.5 %), after an episode of GI bleeding (36.6 %), because of hypersplenism (5.2 %), by chance in the context of other investigations (16.3 %; personal data, unpublished).
The pathogenesis of PH in EHPVO is closely related to the portal vein obstruction, which causes an increased vascular resistance in the portal venous system. Initially, the occlusion of the portal vein by thrombus formation is followed by compensatory vasodilation of the hepatic artery buffering the need for blood supply to the liver. Eventually, collateral venous vessels bypassing the thrombus develop and constitute the so-called “cavernomatous transformation” or “portal cavernoma.” Part of these collaterals may reperfuse the liver, whereas the majority contributes to the porto-systemic shunting developing at various levels in the portal system. Liver tests are usually normal since there is no parenchymal disease apart from mild vascular changes, such as portal venous dilatation and sclerosis.
The management of EHPVO is mainly directed to the treatment of PH complications through medical and endoscopic means. Despite their efficacy in obliterating EV , endoscopic methods have no effect on portal pressure. Conversely, surgical procedures may decompress the portal venous system and normalize the portal vein pressure. Possible indications for surgical treatment include acute variceal bleeding that cannot be controlled by endoscopic means, persistent EV formation, massive symptomatic splenomegaly, growth retardation, and symptomatic portal biliopathy [23, 24]. EHPVO is dealt with more extensively in the non-cirrhotic PH section.
Posthepatic causes of increased resistance to flow are those related to vascular and/or cardiac diseases, including thrombosis/stenosis of the hepatic veins or the atrio-caval junction, any condition increasing the right atrial pressure, such as constrictive pericarditis, severe tricuspidal regurgitation , and right side cardiac failure. The postsurgical status of some congenital cardiac malformations, such as the Fontan circulation, result in increased central venous pressure and increased resistance to liver outflow [25]. Unlike prehepatic PH, in which liver function remains often normal overtime, in posthepatic PH, the liver blood stagnation may compromise liver function leading to cirrhosis [26].
Budd–Chiari syndrome (BCS) is one of the most common causes of post-hepatic PH both in adults and children. BCS is characterized by hepatic venous outflow obstruction at any level from the small hepatic veins to the atrio-caval junction, regardless of the cause of obstruction. The acute increase of vascular resistance secondary to the hepatic venous outflow obstruction causes the sudden appearance of PH, whereas the chronic status may lead to cirrhosis [27].
A wide variety of predisposing causes may determine the onset of the BCS, including congenital or acquired webs of the IVC and thrombotic, inflammatory, or neoplastic processes.
BCS is relatively rare in children. In studies including patients younger than 10 years with PH, the percentage of cases of BCS accounted for 1–7 % of all, although in some areas such as Africa, India, and China, the rate of pediatric BCS may raise up to 16 % [28, 29].
The presentation of BCS can be acute, chronic, or fulminant. In the early course of the disease, it may be asymptomatic and accompanied by normal liver tests. Eventually, the hepatic venous outflow obstruction may lead to hepatic dysfunction associated with abdominal pain, ascites, and hepatosplenomegaly.
Due to its rarity, BCS in children is often diagnosed with some delay. The stage of the disease at diagnosis influences the management strategy and an early diagnosis offers the best chance of cure without major surgery [30].
The management of BCS in pediatric patients may include the use of anticoagulation, thrombolytic therapy and angioplasty with or without stenting, transjugular intrahepatic porto-systemic shunts (TIPS), and, rarely, surgical portosystemic shunts, the latter carrying high risk of thrombotic obstruction. Some patients may end up with end-stage liver disease and require transplantation.
Intrahepatic Causes of PH
Intrahepatic causes of increased vascular resistance have a more various and complicated pathogenesis compared to the extrahepatic forms, and can be further subdivided, according to the relation with the sinusoidal bed, into three subgroups: presinusoidal, sinusoidal, and postsinusoidal.
Presinusoidal venous block can be caused by many conditions, as detailed in Table 68.1. These disorders cause an elevated portal venous pressure, which cannot be detected by the hepatic vein catheter study, since wedge hepatic venous pressure (WHVP) reflects that of the sinusoids that are distal to the lesion, and therefore have normal blood pressure in this condition. Thus, the only useful technique to gather information on the degree of presinusoidal PH is that of the direct measurement of portal or splenic pulp pressure (Table 68.2).
Table 68.2
Hepatic venous pressures according to the pathophysiology of portal hypertension
Etiology of PH | ISP | PVP | RAP | WHVP | FHVP | HVPG | |
|---|---|---|---|---|---|---|---|
Prehepatic | ↑↑ | ↑↑ | N | N | N | N | |
Intrahepatic | Presinusoidal | ↑↑ | ↑↑ | N | N or ↑ | N | N or ↑ |
Sinusoidal | ↑↑ | ↑↑ | N | ↑↑ | N | ↑↑ | |
Postsinusoidal | ↑↑ | ↑↑ | N | ↑↑ | N | ↑↑ | |
Posthepatic | ↑↑ | ↑↑ | N or ↑ | ↑↑ | ↑↑ | N or ↑ | |
Schistosomiasis is one of the leading causes of PH in the developing countries. Liver involvement due to schistosomiasis is caused by one of the two trematode flukes schistosoma mansoni and japonicum. While the former is seen predominantly in Africa and South America, the latter is common in eastern Asia, especially mainland China [31]. The pathogenesis of liver disease here is secondary to entrapment of eggs in the portal venules that cause granulomatous inflammation leading to fibrosis and, in 4–8 % of cases, presinusoidal PH. Portal tract inflammation results from the host response to the parasitic egg in the hepatic venule. The natural history of PH in this condition is closely related to the number of eggs deposited in the liver [32, 33].
Sinusoidal obstruction is mainly due to cirrhosis. It is marked by an increase of HVPG, normal free hepatic venous pressure (FHVP), and raised WHVP (Table 68.2). In sinusoidal PH, WHVP is equal to portal venous pressure because disrupted intersinusoidal communications diminish compressibility and compliance of the sinusoids, allowing direct transmission of portal pressure to the WHVP [34, 35]. In cirrhosis, the increase of vascular resistance occurs at the level of the hepatic microcirculation (sinusoids), and it is secondary to both a mechanical and a dynamic factor. The mechanical factor is represented by the hepatic architectural derangement, and is characterized by hepatocyte swelling, hyperplasia, portal tract inflammation, and fibrosis in response to liver injury. Besides, collagen deposition in the space of Disse may contribute to increased intrahepatic resistance [36]. The dynamic factor is represented by the active contraction of myofibroblasts and vascular smooth-muscle cells of the intrahepatic veins, and it may be modified by endogenous molecules and pharmacological agents, which affect the intrahepatic vascular resistance. Factors that increase the hepatic vascular resistance include endothelin-1 (ET-1), the alpha-adrenergic stimulus, and angiotensin II. Those decreasing hepatic vascular resistance include NO, prostacyclin, and vasodilating drugs (e.g., organic nitrates, adrenolytics, calcium channel blockers) [15, 37, 38].
Among these endogenous factors, ET-1 and NO play a key role in regulating the hepatic vascular resistance. ET-1 is a powerful vasoconstrictor synthesized by sinusoidal endothelial cells that has been implicated in the increased hepatic vascular resistance of cirrhosis, and in the development of liver fibrosis. NO is a powerful vasodilator substance that is also synthesized by sinusoidal endothelial cells. In the cirrhotic liver, the production of NO is decreased, whereas that of ET-1 is increased. The result of these changes is a net vasoconstrictive effect that, in cirrhosis, accounts for approximately 20–30 % of the increased intrahepatic resistance [39–41].
Another dynamic factor that can lead to an increase of intrahepatic vascular resistance is mediated by stellate cells. Hepatic stellate cells (HSCs) are located in the perisinusoidal space of Disse, behind the endothelial barrier, resulting in 5–8 % of all human liver cells and 13 % of sinusoidal cells. HSCs are involved in vitamin A storage and the synthesis of extracellular matrix components, matrix degrading metalloproteinase, cytokines, and growth factors [42].
HSCs have the capacity to contract or relax in response to vasoactive mediators, such as ET-1 and NO, therefore having a crucial role in controlling intrahepatic vascular resistance and blood flow at sinusoidal level. Indeed, stellate cells become “activated” in response to acute or chronic noxae damaging the liver parenchyma, acquiring a myofibroblast-like phenotype. During HSCs activation, their production of extracellular matrix changes qualitatively and quantitatively, leading to an increase of intravascular resistance.
In summary, in cirrhosis, the increased intrahepatic vascular resistance consists of two main components. The “mechanical factor” is fixed and caused by the structural changes, which occur in patients with chronic liver disease mainly in the form of fibrosis and nodule formation [43, 44]. The “dynamic factor” is variable and caused by endogenous mediators (ET-1 and NO) as well as HSCs activation. The main target of the management of PH is represented by medical therapy directed against the “dynamic factor” to decrease the intrahepatic vascular resistance [45, 46].
Postsinusoidal obstruction includes right-sided heart failure, IVC obstruction, small venules BCS , veno-occlusive disease (VOD) . In this setting, WHVP is elevated, whereas HVPG and FHVP can be either normal or elevated, depending on the site of obstruction, intrahepatic postsinusoidal or posthepatic, respectively (Table 68.2). Hepatic VOD is a clinical syndrome occurring early after bone marrow transplantation (BMT) as a result of liver damage by pretransplant conditioning, or chemotherapy for solid tumors. Its incidence in the pediatric BMT population is between 22 and 28 %, with an associated mortality of up to 47 % [47, 48]. The pathologic injury initiates in zone 3 of the liver acinum with subendothelial edema of hepatic venules, fibrin deposition, microthrombosis, venular narrowing, and sclerosis, followed by hepatocyte necrosis [49]. The result is a postsinusoidal increased resistance to hepatic venous outflow resulting in acute PH and, in some cases, multiorgan failure [50–52].
Other Pathogenetic Mechanisms of PH
In some conditions, PH can be caused by the increase of portal venous inflow itself. In patients with an artero-venous communications between the splanchnic arteries and the portal venous system, an artero-portal fistula (APF), the portal flow is markedly increased and arterialized, with the consequent development of presinusoidal PH. APF can be acquired or congenital, but the most common causes are hepatic trauma and liver biopsy, and can be asymptomatic or manifest with PH. Long-standing APF can lead to severe PH characterized by arterial Doppler signal in the portal vein, reversal of the portal flow, and thickening/narrowing of the extrahepatic portal vein. In this setting, radiological procedures represent the best treatment option to close the artero-venous fistula and restore a normal portal vein flow; although, in the congenital forms, the fistula often reappears through the development of new spontaneous shunt formation [53].
In 1898 Banti described a disorder characterized by splenomegaly and hypersplenism, resulting in PH and anemia in the absence of hematological and liver disease. The actual existence of the condition has been questioned for a long time due to the lack of explanation for the development of splenomegaly, hypersplenism, and PH in these patients. Nowadays, Banti’s syndrome is considered the result of microscopic changes of the portal tract that were not detected at the early stages of its clinical description and corresponding to a group of diseases causing non-cirrhotic PH. Hepatoportal sclerosis (HS) is one of the rare disorder characterized by sclerosis of the intrahepatic portal veins resulting in non-cirrhotic PH. HS in children is uncommon but probably underestimated, and only few case reports have been published so far. The cornerstone of the diagnosis of HS is the histology, characterized by portal fibrosis without evidence of either cirrhosis or nodule formation; portal fibrosis is responsible for the increase in the intrahepatic vascular resistance and PH. Nevertheless, the mechanism leading to portal fibrosis and, in general, the entire phenotype of HS are still not well known [54]. Yilmaz reported on 12 pediatric patients with non-cirrhotic PH. On histology, all patients had HS or intimal fibrous thickening of portal vein and periportal fibrosis, acinar transformation, and regenerative nodules not surrounded by fibrous septa. In some of them, there were also signs compatible with cholestatic disease, including neoductular reaction in seven, mild cholangitis in one, and canalicular bile pigment in one [55].
Systemic Hemodynamic Changes in Portal Hypertension
Increased resistance to portal blood flow is likely to be the “primum movens” in the development of PH; however, a variety of hemodynamic changes contribute to amplify the increased portal venous pressure observed in patients with chronic liver disease .
The hyperdynamic syndrome was first described in the 1950s, when some physicians observed that patients with cirrhosis often showed “warm extremities, cutaneous vascular spiders, wide pulse pressure and capillary pulsations in the nail beds.” In 1953, Kowalski and Abelmann published the first study which demonstrated an increase in cardiac output and a decrease in peripheral vascular resistance in patients with alcohol-induced cirrhosis [56]. The recognition of the dangerous effect of this syndrome on multiple organs, though, was achieved only several years later [57].
Vasodilatation plays a key role in the development of the hemodynamic changes. The hyperdynamic syndrome should be better called “progressive vasodilatatory syndrome,” because vasodilatation is the main factor that brings about all the vascular changes and finally the multiorgan involvement seen in cirrhosis [58]. A major step ahead in this field was accomplished in the 1990s, when researchers discovered that NO was responsible for the vasodilatation and, in turn, of the multiple organ malfunctions characterizing the hyperdynamic circulation [59].
Both clinical studies and animal models have demonstrated and explained the hemodynamic events that occur in PH but, since they have not been performed in children, the findings should be interpreted with caution (Fig. 68.2) .
Splanchnic Circulation
Vasodilation of the splanchnic circulation is a process mediated by humoral vasodilatatory agents, and it is probably the initial signal triggering the hyperdynamic systemic circulation. Splanchnic vasodilation causes, as a consequence, an increased portal venous blood inflow, contributing to the maintenance and the aggravation of PH [57, 60]. The result of this significant vasodilation is that a large proportion of circulating blood volume remains confined to the splanchnic system, with a subsequent reduction of central blood volume. This process is called the “forward flow” theory and provides a rationale for the use of vasoconstrictors in adult patients with PH [12].
Systemic Circulation
Splanchnic vasodilation is associated with changes in the systemic circulation, such as a decrease of arterial pressure, that is consequence of the decreased central blood volume and peripheral resistance in various organs [61]. Compensatory mechanisms include the activation of baro- and volume receptors as well as the production of neurohormonal substances leading to sodium and water retention, with plasma volume expansion and increase in cardiac output [62].
The cardiac response is directly related to splanchnic vasodilatation and plasma volume expansion, together with an increased venous return that is mostly due to the formation of porto-systemic shunts. Although vasodilatation is essential as the initiating factor, no hyperdynamic circulation occurs without expansion of plasma volume and porto-systemic shunting [63]. The former is due to renal sodium retention, which has been shown to precede the increase in cardiac output, and can be prevented or reversed by sodium restriction and administration of spironolactone. The latter is characterized by the development of new veins (called collateral vessels) bypassing the liver and decompressing the portal venous system. These veins directly connect the portal blood vessels to veins that divert the blood away from the liver into the systemic circulation. The drawback in this compensatory process is that substances (such as ammonia and toxins) that are normally removed from the blood by the liver, pass directly into the systemic circulation, and have adverse effects in other organs [64].
Collateral vessels tend to develop at the lower end of the esophagus and at the upper part of the stomach (Fig. 68.1). Here, the vessels enlarge and become full of twists and turns, becoming varicose veins in the esophagus (EV) or stomach (gastric varices). Other collateral vessels may develop on the abdominal wall and in the rectum. These vessels are prone to rupture, leading to GI bleeding.
Lung Circulation
PH and liver shunting may also affect the lungs, resulting in the development of hepatopulmonary syndrome (HPS) and porto-pulmunary hypertension (PPH); these conditions are characterized by hypoxia due to pulmonary artero-venous shunts and pulmonary hypertension, respectively [65]. Although the intrinsic mechanism triggering these complications is not fully known, the major role seems to be played by molecules active on the pulmonary endothelium (including NO and carbon monoxide) that can cause either condition [66, 67].
Renal Circulation
Renal circulation is affected indirectly by the hyperdynamic state. To balance the progressive systemic vasodilation, the kidney responds to a perceived hypovolemia by retaining sodium and water. The relative hypovolemia results from an increase of the vascular compartment caused by vasodilatation, leading to the activation of vasoconstrictive and volume-retaining neurohumoral substances that perpetuate sodium and water retention [68]. These compensatory mechanisms include the activation of renin–angiotensin–aldosterone system and antidiuretic hormone secretion. In the early course of the disease, the intravascular volume and the cardiac output increase to maintain the arterial perfusion pressure [69]. With the progression of the disease, vasodilatation worsens, and the cardiac output continues to increase up to a maximum, and then it is not enough to maintain the perfusion pressure. At this point, the renal blood flow drops and renal failure develops [70, 71].
Clinical Manifestation of Portal Hypertension
PH in children has a broad spectrum of clinical manifestations, varying from the occasional finding of splenomegaly discovered during a routine follow-up visit in absence of any symptom, to hematemesis and melena due to the rupture of EV (Table 68.3). The main manifestations of PH are GH, ascites , and splenomegaly, but, in a minority of patients, other complications may arise including hepatic encephalopathy (HE), pulmonary vascular disorders, and kidney disease [72].
Table 68.3
Clinical evaluation and investigations useful to recognize patients with suspected portal hypertension
Step | Aim |
|---|---|
Clinical history | Ask for neonatal umbilical catheterization, episodes of gastrointestinal bleeding, results of previous blood tests, investigations for an undefined splenomegaly |
Physical examination | Assess liver size and consistency, look for splenomegaly, abdominal venous patterning (site and direction of venous flow), spider naevi and telangectasias, palmar erythema, ascites, limbs edema |
Liver function tests | Assess liver function and full blood count for hypersplenism |
Ultrasonography and Doppler of the liver | Evaluate liver parenchyma, patency of portal vein and direction of venous blood flow, hepatic veins patency, venous anatomical abnormalities, hepatic artery (patency and abnormalities), porto-systemic shunts, ascites, splenomegaly, renal abnormalities |
Upper endoscopy | Assess varices and hypertensive gastropathy |
CT scan of the abdomen | Assess liver parenchyma, biliary tree conformation, vascular anatomy, Rex recessus patency and signs of portal hypertensive biliopathy |
Measure portal venous pressure (HVPG, WHVP, FHVP) | Evaluate the degree of portal hypertension. Diagnose prehepatic, intrahepatic, posthepatic causes |
Liver biopsy | Assess fibrosis/cirrhosis, inflammation, histological pattern |
Gastrointestinal Hemorrhage
GH is defined as bleeding in the digestive tract, and it can be classified as proximal or distal, acute or chronic. Bleeding from the upper digestive tract (esophagus, stomach, and upper portion of the small intestine) causes hematemesis and melena, whereas bleeding from the lower digestive tract (lower portion of the small intestine, large intestine, and rectum) causes dark blood or bright red blood mixed with stool, depending on the proximity to the anal sphincter. GH is mainly related to bleeding from EV and also, in a minority of cases, from portal hypertensive gastropathy, gastric antral vascular ectasia, or gastric, duodenal, peristomal, or rectal varices (Fig. 68.3).
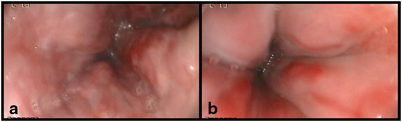
Fig. 68.3
Endoscopic appearance of large esophageal varices with red signs in two children with portal hypertension. (Reprinted from Ref. [73], with permission from Elsevier)
Acute GH is often the first symptom of a long-standing silent liver disease, and therefore it is regarded by patients and carers as a frightening event, giving the impression of imminent death. Although the mortality from GI bleeding in children is lower than in adults, acute GH remains a life-threatening event and requires prompt medical intervention. Chronic bleeding is usually mild and can be discovered since the patient has refractory iron-deficiency anemia and positive fecal occult blood test [73].
The formation of varices and their rupture result from the increased pressure within the vessel as a consequence of PH. When the wall tension exceeds the variceal wall strength, the rupture of the varix occurs, and the patient develops hematemesis and/or melena [74] .
Variceal bleeding in children with chronic liver disease often follows an acute upper respiratory tract infection, with the contribution of several factors such as the increased abdominal pressure during coughing or sneezing, the increased cardiac output due to fever, and the erosive effect of nonsteroidal anti-inflammatory drugs used to treat the fever. Gastroesophageal reflux is another factor which may contribute to erosions of varices leading to its rupture and bleeding [75–77].
Hematemesis and melena are the most common presenting symptoms in children with both intrahepatic and extrahepatic PH, and the first episode can be as early as 2 months of age [2, 22, 78–80].
The age at the first bleeding episode is related to the underlying etiology. In children with biliary atresia, the first bleed was described at a mean age of 3 years, while in children with cirrhosis due to cystic fibrosis it occurred at 11.5 years [74, 81]. In a recent study, 65 children with EHPVO were followed for a median period of time of 8.4 years. Thirty-two (49 %) patients presented with bleeding at a median age of 3.8 years (0.5–15.5) and, during the follow-up period, 43 of them (66 %) had at least one bleeding episode [2]. Triger et al. followed 44 children with EHPVO for a mean follow-up of 8 years. The actuarial probability of bleeding was 49 % at age 16 years and 76 % at 24 years of age. If the child bled before 12 years of age, the probability of bleeding was higher than in those who had not bled before 12 years of age. Further, there was no evidence of variceal regression over time. These studies do not support the previous hypothesis that variceal bleeding decreased in adolescence due to the development of spontaneous porto-systemic collaterals [2, 82]. In a multicenter Italian study on 187 children with EHPVO, the mean age at diagnosis was 4 years, and the most common symptoms at onset were splenomegaly (39.5 %) and bleeding (36.6 %). In 71 patients with an available endoscopy at presentation, 62 (87.3 %) had already developed EV . Development of EHPVO was strictly associated with a neonatal disorder including history of prematurity, neonatal illness, and umbilical venous catheter. Authors concluded that a liver Doppler ultrasound should be performed before discharge from the neonatal unit and at the follow-up to allow an early recognition of the disease and avoid bleeding from EV that are present from the early stages (personal, unpublished data). Since splenomegaly is a very common sign detected in children with PH at the time of GI bleeding, the association between GI bleeding and splenomegaly should be suggestive of PH until proven otherwise [74].
There is no strong evidence supporting the efficacy of any treatment for the prevention of variceal bleeding in children. The administration of nonselective β-blockers (NSBBs), the endoscopic treatment of varices, and the surgical (meso-rex bypass, porto-systemic shunts) and radiological (TIPS) measures to decompress the portal system represent the main therapeutic options for the primary and secondary prophylaxis of bleeding in children with PH [73, 83].
Splenomegaly
Splenomegaly indicates an enlargement of the spleen usually associated with an overactivity of the spleen, defined hypersplenism, which leads to premature destruction of blood cells. Splenomegaly is due to PH which causes at the beginning only spleen congestion and eventually tissue hyperplasia and fibrosis. The increase in spleen size is followed by an increase in splenic blood flow, which participates in PH actively congesting the portal system [84]. Together with EV , splenomegaly represents the most common finding in children with PH even though, in asymptomatic children, it is often discovered accidentally during a routine physical examination.
Despite a big spleen is highly suggestive for PH, many children with liver disease and isolated splenomegaly have often a delayed diagnosis. In clinical practice, splenomegaly accompanied by hypersplenism is considered a sign of hematological disorders, leading to a long hematological follow-up (including bone marrow aspiration and biopsy) before asking consultation to a hepatologist. Due to the large spleen, children with PVT often receive a diagnosis of infectious mononucleosis every time they come to clinical attention because of a viral illness, and PH is disclosed only after a bleeding episode.
Liver function tests and Doppler ultrasound are mandatory in healthy children with splenomegaly and hypersplenism to exclude the presence of EHPVO and avoid worthless procedures [74, 85].
Some studies have tried to identify the best noninvasive method to diagnose the presence of EV in children with PH. Platelet count and splenomegaly are usually considered the most reliable parameter to predict the presence di EV. The clinical prediction rule proposed by Gana has high predictive value (area under the receiver operating characteristic; ROC curve 0.80) and is calculated according to the following formula: (0.75 × platelets)/(spleen z score + 5) + 2.5 × albumin [86].
Ascites
Ascites is the accumulation of serous fluid in the peritoneal cavity, and is usually seen in patients with PH due to cirrhosis. Ascites appears when the hydrostatic pressure goes above the osmotic pressure within the hepatic and mesenteric capillaries, and the transfer of fluids from blood vessels to lymphatics overcomes the drainage capacity of the lymphatic system [89] .
Ascites should be analyzed to obtain information on its cause and possible complications. The serum ascites albumin gradient (SAAG) is used to classify ascites into portal and non-portal hypertensive etiologies. The SAAG is calculated by subtracting the ascitic fluid albumin level value from the serum albumin value, and the result correlates directly with portal pressure. This phenomenon is the effect of Starling’s forces between the fluid of the circulatory system and ascitic fluid, as albumin does not move across membranes easily, because it is a large molecule. Under normal circumstances, the SAAG is ≤1.1 g/dl because serum oncotic pressure (pulling fluid back into circulation) is exactly compensated by the serum hydrostatic pressure (which pushes fluid out of the circulatory system). In presence of PH, there is an increase in the hydrostatic pressure causing more fluid and more albumin to move from the circulation into the peritoneal space with ascites formation. As a consequence, the SAAG increases (≥1.1 g/dl). Thus, a high gradient (SAAG ≥1.1 g/dl) indicates that the ascites is due to PH, whereas a low gradient (SAAG ≤1.1 g/dl) indicates that ascites is not associated with increased portal pressure (Table 68.4). In clinical practice, some conditions may influence the proper value of the SAAG including the sampling of ascites and serum in different states of hydration or the impact of serum globulin concentration [90, 91] .
Table 68.4
Causes of ascites based on serum ascites albumin gradient (SAAG)
SAAG ≥1.1 g/dl = portal hypertension | SAAG ≤1.1 g/dl = other causes of ascites |
|---|---|
Cirrhosis | Peritoneal lymphoma |
Non-cirrhotic liver disease | Serositis |
Fulminant hepatic failure | Chronic peritoneal infection |
Tubercolosis | |
Other (bacteria, viruses, fungi) | |
Vascular/heart disease | Low serum colloid osmotic pressure |
Portal vein thrombosis | Nephrotic syndrome |
Veno-occlusive disease | Protein-losing gastroenteropathy |
Budd–Chiari syndrome | Kwashiorkor |
IVC obstruction/right heart failure | Hollow organ leak |
Benign and malignant neoplasms | Lymphatic |
Mixedema | Other (pancreatic, biliary, intestinal) |
Ascites should also be evaluated for spontaneous bacterial peritonitis (SBP), an ascitic fluid infection without an evident intra-abdominal surgically treatable source. The diagnosis is made by ascitic fluid cell count. The absolute polymorphonuclear cell (PMN) count in the ascitic fluid is calculated by multiplying the total white blood cell count (or total “nucleated cell” count) by the percentage of PMNs in the differential. The diagnosis of SBP is established by an elevated ascitic fluid absolute PMN count (≥ 250 cells/mm3), a positive ascitic fluid bacterial culture, and absence of secondary causes of peritonitis [92]. Patients with SBP should receive antibiotic therapy, such as intravenous third-generation cephalosporin, and be considered for liver transplantation.
Treatment of ascites includes salt and fluid restriction and use of diuretics. Spironolactone is the diuretic of choice as it is an aldosterone antagonist counteracting the endocrine changes of the hyperdynamic circulation, but often there is the need to add a loop diuretic, such as furosemide, that can improve diuresis and counteract hyperkalemia. In children with normal liver synthetic and biliary function, ascites can often be managed with diuretics and occasional paracentesis (Fig. 68.4). Paracentesis has been utilized safely in children and is indicated when ascites is large and not responding to diuretics [93, 94]. When ascites does not recede, recurs shortly after paracentesis, or when children do not tolerate diuretic therapy due to side effects, the management can take advantage of more aggressive treatment including regular large-volume paracentesis and, if feasible, TIPS . TIPS procedure, although uncommon in children, provided good results in term of resolution of refractory ascites in both native and transplanted livers [88] .
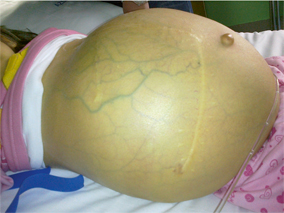
Fig. 68.4
Tense ascites and abdominal venous patterning in a child with biliary atresia, failed Kasai, and end-stage liver disease
When ascites is accompanied by signs of end-stage liver disease, such as hypoalbuminemia , jaundice, clotting derangement, or SBP, the only effective treatment is liver transplantation. In these cases, albumin infusions can be used along with diuretics, in order to increase the osmotic pressure and facilitate the passage of fluid from the extravascular to the intravascular compartment. In children with end-stage liver disease, ascites can be associated with hyponatremia, which is a risk factor for severe complications and death. Pugliese et al. evaluated the association of pretransplant variables with the mortality within 90 days following the inclusion on the waiting list of 520 children with cirrhosis. On multivariate analysis, the presence of ascites and serum sodium levels were associated with decreased patient survival while awaiting a liver graft [95].
Chylous ascites is a rare clinical condition marked by an extravasation into the peritoneal cavity of a milky fluid deriving from the mesenteric lymphatic vessels. Usually, it results from major abdominal surgical interventions, such as liver transplantation, during which several lymphatic vessels are inadvertently resected and PH has not yet resolved; nevertheless, chylous ascites can present also in patients with PH due to PVT or congenital portal venous malformation. In this setting, in spite of the absence of strong evidences, the management includes fat-free diet and somatostatin analogues [96] .
Pulmonary Complications
Children with PH may develop two rare pulmonary complications: hepatopulmonary syndrome (HPS) and portopulmonary hypertension (PPH). Their relative frequency and risk factors have not been defined, and only isolated cases or small series have been published so far.
The pathogenesis of HPS and PPH remains unclear, but the two conditions arise only in patients with porto-systemic shunting, and therefore the pathogenesis must be related to it. The proposed theories suggest that these disorders result from a combination of the hyperdynamic circulation, the increased cardiac output, the sheer injury to the vascular walls, and an imbalance of circulating vasoactive peptides. Abnormal hepatic synthesis of vasoactive peptides, such as EN-1, or impaired hepatic metabolism of intestinally derived endotoxins, cytokines, and neurohormones may result in these substances reaching the pulmonary vascular bed via porto-systemic shunting, directly altering the vessel tone or leading to pulmonary vascular inflammation and remodeling. The resulting pathology is strikingly different in these two disorders, with vasodilation of pulmonary arterioles and capillaries causing artero-venous shunting in HPS, and intimal fibrosis with endothelial and smooth-muscle cell proliferation leading to increased pulmonary vascular resistance in PPH [97–99].
HPS is defined as intrapulmonary vascular shunting (IPVS), ventilation–perfusion mismatch and chronic hypoxemia in a setting of liver disease and/or PH. The mechanisms implicated in the development of HPS are likely to include many of the vasoactive substances involved in the genesis of the hyperdynamic circulation, including NO and EN-1 [100–102]. Porto-systemic shunting plays a key role in the pathogenesis of the HPS; in fact, HPS has been described also in patients with congenital porto-systemic shunting and no liver disease (i.e., the Abernethy malformation).
From the clinical point of view, HPS is characterized by shortness of breath, exercise intolerance, and digital clubbing. Since the disease is often subtle and progresses slowly, in the early stages it can be easily overlooked and become overt only when advanced. Patients with PH should be screened for HPS by measuring the transcutaneous oxygen saturation and, if < 96 %, by performing further investigations to assess the real presence of IPVS. The two main procedures to confirm the presence of IPVS are the echocardiography with agitated saline and the macroaggregated albumin scan. The former is simple and sensitive in both symptomatic and asymptomatic children. The latter may be used to quantify the degree of shunting, which can be useful in clinical decision-making and to test the progression of HPS over time [103–105].
Liver transplantation represents the only effective treatment option for children with HPS. Al-Hussaini et al. reported a study on 18 children with HPS over 14 years. Fourteen underwent LTX with resolution of HPS in 13. Six developed vascular or biliary complications and four died (two before transplantation) [106].
PPH is defined as an elevation of the mean pulmonary arterial pressure and increased vascular resistance caused by a pulmonary arteriopathy, in the setting of PH and in the absence of underlying cardiopulmonary disease [105]. The pathophysiology of this disorder is still unclear; however, it seems to be related to a decreased hepatic clearance and porto-systemic shunting of biochemical mediators in the setting of liver dysfunction and PH. PPH produces characteristic histological changes in the pulmonary vasculature that have been well documented on autoptic samples [107].
Although there are scant data in children, it has been shown that PPH can develop in patients with cirrhotic and non-cirrhotic causes of PH such as biliary atresia, PVT, focal nodular hyperplasia, and congenital hepatic fibrosis [108]. From the clinical point of view, PPH presents most commonly with exertional dyspnea, fatigue, palpitations, and syncope or chest pain. The symptoms are often subtle at onset, so that a high index of suspicion is required to diagnose PPH in asymptomatic patients before they develop severe and irreversible pulmonary hypertension [107]. Echocardiography with Doppler flow is considered the most useful screening modality in patients with suspected pulmonary hypertension, although limited by the fact that it relies on the presence of tricuspidal regurgitation , it measures systolic rather than mean pressure in the right ventricle and the measurement is approximate. Chest X-ray and electrocardiogram (ECG) definitely lack sensitivity. If there are indirect signs of pulmonary hypertension at ECG, further evaluation should include a right heart catheter study and, in some cases, high-resolution computed tomography (CT) of the chest and ventilation-perfusion lung scanning [104].
If the diagnosis of PPH is made early before the development of irreversible pulmonary vasculopathy, LTX can be successfully performed and may reverse the process. Conversely, once PPH is advanced (with a mean pulmonary pressure > 35 mmHg) and associated with right-sided heart failure, LTX becomes unfeasible because of the functionally obstructed liver outflow that leads to graft failure and death in at least 50 % of cases. More recently, it has been shown that the perioperative use of inhaled and intravenous pulmonary vasodilators (NO and epoprostenol) as well as oral drugs (sildenafil and bosentan) can remarkably reduce the pulmonary pressure to a safe level, allowing to perform LTX [108]. The goal of treatment is, therefore, lowering the mean pulmonary arterial pressure and move the patient from high risk to a safer ground for transplantation. However, if there is no response or the pressures remain very high, the only viable option is a combined lung–liver transplant.
Other Major Complications of Portal Hypertension
An abnormal abdominal venous patterning can be seen in children with PH, in whom a prominent subcutaneous vascular pattern develops as part of spontaneous porto-collateral shunting (Fig. 68.4). This is the result of the attempt at decompressing the portal venous system through the umbilical vein recanalization that leads to periumbilical collaterals. Although less common than in adults, both umbilical venous shunts and rectal varices can be observed in children with long-standing PH, whereas in children with PH and an intestinal stoma (i.e., in short bowel syndrome associated with liver disease), stomal varices often occur and represent a site of low resistance and bleeding [109].
Hepatorenal syndrome (HRS) is defined as a functional renal failure in patients with liver disease and PH, and it constitutes the climax of the systemic circulatory changes associated with PH [91]. In pediatric patients, HRS is rare, probably due to the relatively short time that cirrhotic children spend on the transplantation waiting list. Two types of HRS have been identified. Type 1 HRS is an acute and rapidly progressive form that often develops after a precipitating factor such as GI bleeding or SBP. Type 2 HRS is a slowly progressive form of renal failure that often occurs without a sudden trigger in the setting of chronic and refractory ascites . HRS arises from severe vasoconstriction of the renal circulation to compensate for the characteristic circulatory imbalance of advanced cirrhosis. This leads to an increased renal arterial resistance which in turn causes renal hypoperfusion and arterial hypotension. The small volume of the produced ultrafiltrate is then reabsorbed almost completely in the proximal tubule, whereas no solutes (such as sodium) flow to the Henle’s loop with nearly no hyperosmolar natriuresis, activation of adiuretin–vasopressin and reduced urine output. As a result, standard diuretic treatment has little effect on diuresis [110]. The criteria to diagnose HRS are difficult to be applied in young children because of the lack of pediatric data. HRS is a potentially reversible condition, but its natural prognosis is poor. Various vasoconstrictors are useful in the treatment of HRS, and terlipressin is the first choice [111]. In the pediatric setting, the experience is little. In a report, four children with end-stage liver disease received terlipressin treatment for renal failure compatible with HRS type 1 in three and type 2 in one. All four responded well and no side effects were reported [112]. Liver transplantation is the ultimate treatment for HRS, ensuring full recovery and long-term survival, and thus it remains the principal tool both in adults and children.
Hepatic encephalopathy (HE) refers to a variety of reversible neurological abnormalities reported in patients with cirrhosis and PH associated with anatomical and functional porto-systemic shunting. In children, HE can be subtle, and the condition seems to appear at a later stage of liver disease and be difficult to diagnose, especially in ill infants. Disturbed consciousness (including coma), personality changes, intellectual deterioration, and speech and motor dysfunction are common in older children with HE. These symptoms usually have a sudden onset and a rapid reversibility suggesting they are of metabolic origin [113].
Non-cirrhotic Portal Hypertension
Non-cirrhotic PH (NCPH) is a heterogeneous group of liver disorders characterized by PH in absence of cirrhosis and with normal or only mildly elevated HVPG values [116].
They are of crucial importance in pediatric hepatology since, while the majority of children with cirrhotic PH are treated with liver transplantation successfully in the early years of the life, those with NCPH do not have any indication for LTX and are managed and followed up for a long time up to adult age [73].
In relation to the site of increased vascular resistance to blood flow, such disorders may be classified as prehepatic, hepatic, and posthepatic. Among presinusoidal NCPH disorders, non-cirrhotic portal fibrosis (NCPF) and EHPVO represent two different entities in whom features of PH are not associated to significant parenchymal dysfunction [116].
NCPF is mostly a disorder of young adults or middle-aged women, whereas non-cirrhotic EHPVO is reported both in infancy and in older children. Recently, it has been proposed the so-called unifying hypothesis, providing a common explanation to the pathogenesis of both NCPF and EHPVO, and focusing on thrombotic events affecting the portal branches. The authors hypothesize that a major thrombotic event occurring at early ages and involving the portal trunk results in EHPVO, whereas repeated microthrombotic events occurring later in life and affecting the small or medium branches of the portal vein would lead to NCPF [117]. In this session, we focus on such two disorders. NCPF is a rather unknown liver disorder in children, whereas EHPVO represents the most common cause of NCPH in the pediatric population.
Non-cirrhotic Portal Fibrosis (NCPF)
The clinical pattern of presentation of NCPF is that of PH in the absence of an evident cause, such as liver fibrosis/cirrhosis or vascular obstruction. NCPF is also named idiopathic PH (IPH), idiopathic non-cirrhotic portal hypertension (INCPH), hepatoportal sclerosis (HS), and obliterative venopathy [116].
On histology, the main features include phlebosclerosis, fibroelastosis, periportal and perisinusoidal fibrosis, aberrant vessels in portal tract (portal angiomatosis) with preserved lobular architecture, and differential atrophy. The main portal vein branch is dilated, with thick sclerosed walls, along with thrombosis in the medium and small portal vein branches, giving a picture of “obliterative portal venopathy” [118, 119]. However, in children, these features are often subtle and the condition may be overlooked.
The etiology of NCPF is undefined, but the attention has been brought to various factors that may trigger autoimmunity or endotoxin-mediated injury leading to vascular abnormalities, which cause presinusoidal block to the portal venous flow. The interest has been pointed particularly on lack of hygienic conditions, that would support the role of infections as trigger of the disease, and prothrombotic disorders, that would support the association with an underlying prothrombotic state [120].
The diagnosis of NCPF is based on clinical evidence of PH without liver dysfunction and a histology with no significant fibrosis. The Asian Pacific Association for the study of the liver (APASL) has proposed some criteria for the diagnosis of NCPF in adults [121]. Recently, Schouten et al. redefined five criteria including:
1.
Any one of the following clinical signs of PH: splenomegaly, EV, ascites, raised HVPG, and evidence of porto-systemic collaterals
2.
Exclusion of cirrhosis on liver biopsy
3.
Exclusion of known causes of chronic liver disease causing cirrhotic or non-cirrhotic PH
4.
Exclusion of common conditions causing NCPH
5.
Patent portal and hepatic veins
All five criteria must be fulfilled to diagnose NCPF [122].
In adults, the most common symptoms at onset include bleeding from varix rupture, splenomegaly with or without hypersplenism, and ascites in 10–34 % of the cases. On physical examination, the liver may be normal, enlarged, or slightly shrunken, whereas the clinical signs of chronic liver disease are absent [123]. Liver function tests are usually normal in NCPF, but derangements in liver enzymes, prothrombin time, and albumin are seen in a small proportion of adult patients [124].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


