Small-intestinal enteropathy of unknown origin
Intractable ulcerating enterocolitis of infancy
Congenital enterocyte heparan sulfate deficiency
Congenital intestinal integrin deficiency
Congenital secretory diarrheas
Congenital chloridorrhea
Congenital Na-losing diarrhea
Autoimmune enteropathy
Diseases of the intestinal epithelium
Microvillus inclusion disease
Tufting enteropathy
Autoimmune Enteropathy
This rare disorder mostly occurring in young infants and children (6–18 months old), is characterized by severe diarrhea and small-intestinal mucosal atrophy resulting from immune-mediated injury. It remains a challenging diagnosis because of its clinicopathologic variability. This entity is dealt with in Chap. 2.
Small-Intestinal Enteropathy of Unknown Origin
This entity could be a variation of autoimmune enteropathy, as the increase in inflammatory cells in the lamina propria shows. It appears in less than 12-month-old infants, with a lower death rate compared to those with autoimmune enteropathy, but it can be very severe. Infants can be dependent from TPN [5].
Intractable Ulcerating Enterocolitis of Infancy
A rare disease initially described in 1991 in five children presenting in the first year of life with intractable diarrhea, ulcerating stomatitis, and large ulcers with overhanging edges throughout the colon within the first year of life [7]. The affected infants can show such a severe colitis that a subtotal colectomy is necessary, even if long-term prognosis is good. It has been suggested that the affected children have a genetically determined primary immune dysregulation [8].
Congenital Enterocyte Heparan Sulfate Deficiency
Described in 1995 in three infants who, within the first weeks of life, presented secretory diarrhea and massive enteric protein loss [9]. The small-intestinal mucosa is normal on light microscopy, but histochemical examinations show a complete absence of enterocyte heparan sulfate. The sulfated glycosaminoglycans of the basocellular membrane are mostly deficient, particularly heparan sulfate, while distribution of vascular and lamina propria glycosaminoglycans is normal [9]. Diarrhea is so severe to make TPN necessary, associated to repeated albumin infusions because of severe protein-losing enteropathy. Studies in men and mice show that heparan sulfate is essential in maintaining intestinal epithelial barrier function [10], and that the specific loss of heparan sulfate proteoglycans from the basolateral surface of intestinal epithelial cells is common to many forms of protein-losing enteropathy [11].
Congenital Intestinal Integrin Deficiency
In 1999, Lachaux et al. described an intractable diarrhea starting from 9 days after birth, associated to pyloric atresia and total epithelial detachment of gastric and intestinal mucosa. Immunofluorescence analysis showed α6β4 integrin deficiency at the intestinal epithelium—lamina propria junction [12].
Mutations in α6 or β4 integrins cause junctional epidermolysis bullosa with pyloric atresia. In 2008, two Kuwaitian brothers with pyloric atresia were described, respectively affected by intractable diarrhea and episodes of protein-losing enteropathy, with a novel mutation in β4 integrin not associated to its reduced expression in tissues [13].
Congenital Secretory Diarrheas
Includes congenital chloridorrhea and congenital sodium diarrhea, dealt with in Chap. 36.
Diseases of the Intestinal Epithelium
Microvillus inclusion disease and tuft enteropathy are the best-known diseases of the intestinal epithelium causing intractable diarrhea of infancy .
In 1994, Girault et al. described eight infants with early-onset severe watery diarrhea associated to facial deformities and unusual tufts of woolly hair with trichorrhexis nodosa. Duodenal biopsies showed moderate to severe villous atrophy, with normal or hypoplastic crypts; colon biopsies were grossly normal. As a consequence, severe malabsorption was present. All patients had no antibody response to immunization antigens; the immunological response to vaccinations was poor. Five children died despite TPN [14]. Two children from the series of Girault et al. had hepatic cirrhosis; six additional patients had signs and symptoms compatible with this new “syndromic diarrhea”, associated to hepatic involvement (Tricho-Hepato-Enteric Syndrome, THES) characterized by fibrotic livers with marked hemosiderosis [15–17].
Nine different mutations in TTC37 gene (5q14.3–5q21.2) were found in 12 children from 11 families with classical features of THES. TTC37 codes for a protein that has been named thespin (THES ProteIN) [18].
Enlarged platelets with abnormal α-granule secretion can be observed in some patients. The estimated incidence of the syndrome is 1 in 400,000 to 1 in 500,000 live births .
Microvillus Inclusion Disease
In 1978, Davidson et al. described five infants presenting an intractable diarrhea of infancy characterized by secretive diarrhea and malabsorption , starting in the first hours after birth with hypoplastic villous atrophy in the small-intestinal biopsy . Four of these infants had a deceased brother who had presented similar features.
In one of these infants, the electron microscopy identified the presence of a peculiar abnormality of the microvilli of the enterocytes [19] (Fig. 1.1).
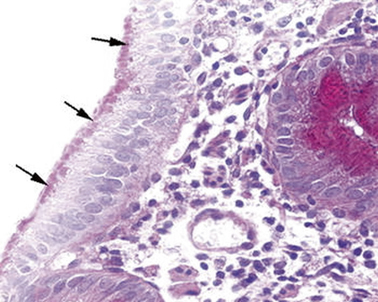
Fig. 1.1
Microvillous in the original label inclusion disease. PAS staining highlights abundant PAS-positive material (arrows) in the apical part of the enterocyte cytoplasm. PAS × 260. PAS peroidic acid-Schiff (Reprinted from Ref. [20], Fig. 1, with kind permission from Springer Science and Business Media)
Three new cases with the same clinic and histological characteristics of the latter were described in France in 1982, and the four of them were grouped in a new disease called congenital microvillus atrophy [21, 22]. Two new cases were described in Great Britain in 1985 [23] and one in Italy in 1986; a subsequently born brother of the latter resulted affected too [24]. A survey completed in 1987 among centers known for their involvement in pediatric gastroenterology identified more than 30 cases worldwide. Additional cases were later published.
In 1989, Cutz et al. proposed the use of the term “microvillus inclusion disease” to highlight the characteristic ultrastructural lesions of the disease [25].
Clinical presentation: case report
First child of parents with no blood relation, A.G. was born after 37 weeks of gestation, the pregnancy having been complicated by a risk of miscarriage in the 5th month. His weight was 3500 g.
The infant was hospitalized when he was 40 days old because of an abundant diarrhea (15–20 evacuations a day of liquid stools), starting on the 6th day of its life and resistant to numerous dietary and pharmacological therapies.
Entering the hospital, the patient weighed 2800 g, it was in severe general conditions with dystrophia and dehydration; a TPN was therefore immediately started. The acid–basic balance showed hyponatremic acidosis (pH 7.2; EB −8,3; Na 128 mEq/l). The secretive nature of diarrhea was confirmed by its entity (about 100 ml/kg/die) with a total absence of oral nutrition and with the persistence of TPN in progress.
Moreover, the typical absence of ionic gap in the stools was present: osmolality 226 mOsm/l, Na 86 mEq/l, K 23.5 mEq/l (gap 7 mOsm/l).
Loperamide and chlorpromazine increased intestinal absorption, but did not change the clinical picture.
Microbiological examinations included an electronic microscope examination of the feces for the identification of viruses and the search for enterotoxigenic bacteria and parasites with specific methods were repeatedly negative.
The abdominal ultrasound showed adrenal hyperplasia associated to hyperaldosteronism (1160 ng/ml, v.n. < 125 ng/ml).
Jejunal biopsy showed a picture of villus atrophy with no hyperplastic crypts and periodic acid-Schiff (PAS)-positive material stored in the apical cytoplasm of enterocytes. Electron microscopy was diagnostic for microvillus inclusion disease.
Microvillus Inclusion Disease is a Congenital Secretory Diarrhea Starting in Neonatal Age
Severe diarrhea typically appears in the first days of life, usually within the first 72 h, and it is immediately life threatening. The stools are watery, and the stool output is 100–500 ml/kg/day when the infant is fed, a volume comparable to or higher than that observed in cholera. The diarrhea is of secretory type; therefore, it persists at a stable rate of 50–300 ml/kg/day despite fasting, and the electrolyte content of the stools is increased, without an osmotic gap. However, the mucosal atrophy causes osmotic diarrhea. For this reason, feeding increases the fecal output and oral feeding in nutritionally significant amounts is impossible. Due to the high output, patients can lose up to 30 % of their body weight within 24 h, resulting in profound metabolic acidosis and severe dehydration, unless vigorous intravenous rehydration is started .
In a small percentage of cases (which was in the past considered to be around 20 %, and presently around 5 % of the cases [26]), diarrhea starts later in life, between 1 and 3 months, usually at 6–8 weeks of age. This less severe form has been denominated late-onset microvillus inclusion disease, while the classical form beginning at neonatal age has been denominated early-onset microvillus inclusion disease[27].
The hallmark of the disease is the electron microscopic finding of disrupted enterocytic microvilli (i.e., digitations of the apical membrane of the intestinal epithelial cell protruding into the lumen) and the appearance of characteristic inclusion vacuoles, whose inner surfaces are lined by typical microvilli. Both lesions are seen only with the electronic microscopy .
A few cases have been termed atypical microvillus inclusion disease, in which the onset can be early or late, but the histologic picture is different, particularly for the absence of detectable microvillus inclusions [28].
Therefore, three variants of the disease have been identified: early-onset microvillus inclusion disease, late-onset microvillus inclusion disease, and atypical microvillus inclusion disease. However, because of the sparse distribution of microvillus inclusions, it is not certain that their absence could be limited to the sample.
Microvillus inclusion disease is usually characterized by growth retardation and some developmental delay later in infancy. No other specific findings can be detected. However, the disease can be associated with other abnormalities, indicated in Table 1.2.
Table 1.2
Anomalies described in association to microvillus inclusion disease
Meckel diverticula | Abdominal adhesions |
Inguinal hernias | Renal dysplasia |
Absent corpus callosum | Hydronephrosis |
Mesenteric duct remnants | Craniosynostosis |
Abnormal vertebrae | Down syndrome |
Aganglionic megacolon | Hematuria |
Pneumocystis jiroveci pneumonia | Dihydropyrimidinase deficiency |
Autosomal dominant hypochondroplasia | Microcephaly |
Renal Fanconi syndrome | Other renal problems |
Hypophosphatemic rickets | Diabetes |
Cardiac problems | Pulmonary problems |
Liver dysfunction | Multiple hepatic adenomas |
Histologic Findings
Findings from duodenal biopsy must not be considered diagnostic. Histologic results of duodenal biopsy samples can range from essentially normal to mildly abnormal, showing the following:
Thin mucosa caused by hypoplastic villus atrophy
Diffuse villus atrophy (loss of villus height)
Crypt hypoplasia
PAS staining of the intestinal biopsy sample does not show the usual linear staining along the brush border, but reveals PAS-positive material in the apical cytoplasm. The PAS staining material corresponds to the increased number of electron dense secretory granules in the epithelium. The abnormal pattern of staining appears in the upper crypt region and continues over the villus [29] (Fig. 1.2).
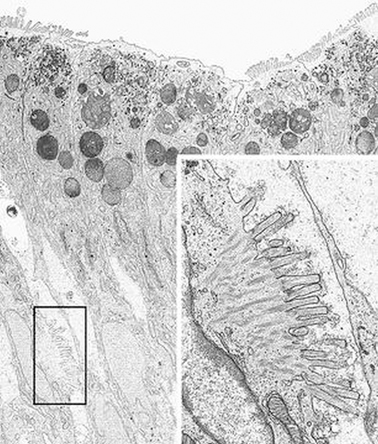
Fig. 1.2
Microvillous in the original label inclusion disease. Villous enterocytes: the boxed area shows microvilli on the lateral membrane. Inset: Enlargement of the boxed area × 6200, inset × 22,500. (Reprinted from [20, Fig. 5], with kind permission from Springer Science and Business Media)
PAS accumulates in low crypts in atypical microvillus atrophy, in upper crypts in congenital microvillus atrophy, and in low villi in late-onset microvillus atrophy.
Similar results were obtained with anti-CD10 immunohistochemistry: In affected children, the normal linear staining in surface enterocytes is absent, while prominent cytoplasmic reactivity is seen [30]. CD10 is a neutral membrane-associated peptidase; thus, abnormal stain findings with PAS or anti-CD10 immunohistochemistry are expressions of the abnormalities in microvillar structure.
Rectal biopsy findings demonstrate microvillus involutions and an increased number of secretory granules. This test has been proposed as a relatively easy method for making an early diagnosis. Anti-CD10 immunohistochemistry can aid in the diagnosis, because abnormal cytoplasmic CD10 staining of absorptive colonocytes has been observed in microvillus inclusion disease [31].
The diagnosis rests on findings demonstrated by electron microscopy (see Figs. 1.3 and 1.4.). Electron microscopy shows well-preserved crypt epithelium with abundant microvilli. Villus enterocytes are severely abnormal, particularly toward the apices of the short villi. The microvilli are depleted in number, short, and irregularly arranged. Some of the enterocytes contain the typical microvillus involutions, which are intracellular vacuoles where microvilli are observed lining the inner surface. A striking feature is a number of small, membrane-bound vesicles containing electron-dense material (see Figs. 1.3 and 1.4). A few cases have been described in which the classic microvillus inclusions are shadowed by other features, such as large aggregates of electron lucent, vermiform membranous vesicles in enterocyte cytoplasm, corresponding to the PAS-positive material [32].
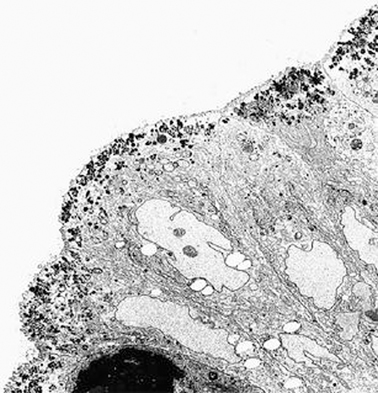
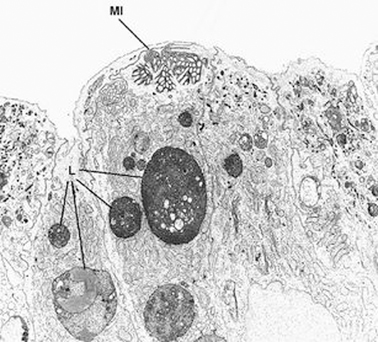
Fig. 1.3
Microvillous in the original label inclusion disease. The apical cytoplasm of villous epithelium shows an increased number of secretory granules associated with microvillus alterations × 2400. (Reprinted from [20, Fig. 4], with kind permission from Springer Science and Business Media)
Fig. 1.4
Microvillous in the original label in the original label inclusion disease. The villous enterocytes lack brush-border microvilli, whereas their apical cytoplasm contains a microvillus inclusion (MI) and numerous lysosomes (L) × 5.500. (Reprinted from [20, Fig. 2], with kind permission from Springer Science and Business Media)
Epidemiology
The cases published or gathered in an online registry were 137 in 2014 [26].
A female preponderance had been observed among the published cases, with a female-to-male ratio of 2:1, but in the total 137 cases there is a 1.54 male to female ratio. A blood relation is present in 41 % of the assessable cases with a genre preference for males. A cluster of cases from the Navajo reservation in northern Arizona suggests an incidence as high as 1 case per 12,000 live births .
Pathophysiology
Due to their alterations, mature enterocytes inefficiently absorb ions and nutrients, causing a malabsorption syndrome; however, the diarrhea is caused mainly by active secretion of water and electrolytes in the intestinal lumen (secretory diarrhea) . The pathogenesis of the secretory diarrhea is unknown; it is assumed to result from an imbalance between decreased absorption and unaltered secretion.
Measurement of stool electrolytes and osmolality enables rapid and accurate assessment of the pathogenesis of this chronic diarrhea (osmolar versus secretory) and greatly narrows the differential diagnosis.
Fecal electrolytes demonstrate a typical pattern of secretory diarrhea. Fecal sodium levels are high (approximately 60–120 mEq/l), and no osmotic gap is found. In patients with secretory diarrhea, the following formula applies: 2(Na concentration + K concentration) = stool osmolarity ± 50. In osmotic diarrhea, stool osmolarity exceeds 2(Na concentration + K concentration) by 100 or more.
Secretory diarrhea occurs in the fasting state and is associated with large output losses that cause dehydration and metabolic acidosis.
In osmotic diarrhea, findings on stool microscopy are negative for white blood cells (WBCs), blood (exudative diarrhea), and fat (steatorrhea).
Even if there are data about the anomalies in water and electrolytes transportation in the small intestine, it is not known whether and how the colon mucosa participates to the absorption alterations in the disease.
In one of the Italian cases, we used the technique of rectal perfusion that showed a decrease in sodium absorption, only partially corrected by chlorpromazine administration [33].
Pathogenesis
Severe perturbation of the microvillar cytoskeleton may disrupt the transport of brush border components that have to be assembled at the apical membrane. The postulated abnormality in the cytoskeleton causes a block in exocytosis, mainly of PAS-positive material (e.g., polysaccharides, glycoproteins, glycolipids, neutral mucopolysaccharides). As a consequence, small secretory granules that contain a PAS-positive material accumulate in the apical cytoplasm of epithelial cells.
In 2008, the presence of mutations in the MYO5B gene was described in seven patients (out of ten tested), predominantly of Turkish origin [34]. Homozygous mutations in the same gene were subsequently found in seven cases of Navajo origin; five parents were heterozygote [35]. A total of 41 unique MYO5B mutations in 40 patients have been identified so far: in more detail 16 different homozygous mutations in 25 patients, and 25 heterozygous mutations in 15 patients [26].
The MYO5B gene codifies myosine Vb, an actin-based motor protein which carries the recycling endosomes to the apical plasma membrane along the actin filaments of the microtubules. The functional deficiency of this protein alters the intracellular trafficking of resident apical plasma membrane proteins to the cell surface, and this could be the cause for the impaired apical brush border membrane development [34]. Actually, in microvillus inclusion disease the MYO5B mutations associate to a defective myosin Vb expression in enterocytes.
Myosine Vb carries on its action after having bound to a specific small guanosine-5′-triphosphatase (GTPase) rab proteins, such as Rab 11, located on the surface of recycling endosomes [36]. Thanks to this link the recycling endosomes move along the actine filaments (see Fig. 1.5) [37].
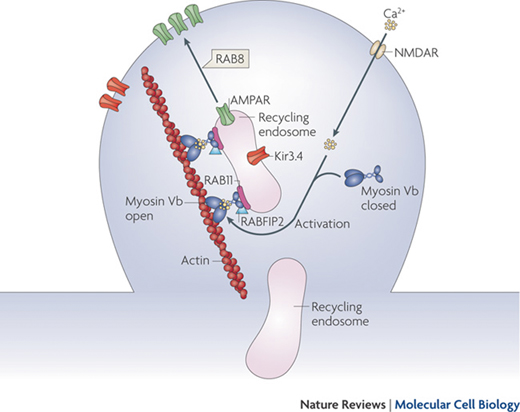




Fig. 1.5
Endocytic recycling. Myosin Vb is a conformation-dependent binding partner of Rab11-FIP2. Activation of myosin Vb induces translocation of recycling endosomes and their cargo. Final transport from the recycling endosome to the cell surface is mediated by Rab8. (Reprinted by permission from Macmillan Publishers Ltd and Nature Publishing Group [37])
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
