Cholestasis
α1-antitrypsin deficiency
Arginase deficiency (UCD)
Bile acid metabolism defects, e.g. CTX
Byler disease
CDGs
Cholesterol synthesis defects
Citrin deficiency
Galactosaemia
LCHAD
Mevalonic aciduria
Niemann–Pick C (LSD)
Peroxisomal defects
Tyrosinemia type 1
Transaldolase deficiency
Wolman disease (LSD)
Acute liver failure
α1-Antitrypsin deficiency
CDGs
Galactosaemia
Hereditary fructose intolerance
Fat oxidation defects (FAOs)
Mitochondrial disorders including Alpers disease
Neonatal hemochromatosis
Organic acidemias (OAs)
Tyrosinemia type 1
Urea cycle defects (UCDs)
Wilson disease
Hepatomegaly (main or isolated symptom)
α1-antitrypsin deficiency
Cholesterol ester storage disease and Wolman disease
Galactosaemia
Fanconi–Bickel syndrome
Glycogen storage disorders (GSD I, III, IV, VI, and IX)
Lysosomal storage disorders (LSDs)
Farber disease
Gaucher disease
Mucolipidoses
Mucopolysccharidoses (MPS)
Niemann–Pick B&C
Neonatal haemochromatosis
Peroxisomal disorders
Tyrosinemia type 1
Wilson disease
Encephalopathy
FAOs
Fructose 1,6 bisphosphatase deficiency
Mitochondrial disorders
OAs
UCDs
Diagnosis
History
Diagnosis is hampered by the rarity of these conditions, non-specific presentation and the requirement for specific investigations. Diagnostic clues are therefore sought from history and examination to suggest the potential for an IMD (see Table 64.2) and remain the foundation of diagnosis [1].
Table 64.2
Clues from the history suggestive of an underlying IMD
Family history | Consanguinity |
Previous sudden infant death | |
Positive family history and inheritance pattern | |
Only males affected suggests X-linked | |
Matrilineal inheritance suggests mitochondrial DNA point mutation | |
Maternal obstetric history (if an infant or childhood presentation) | Multiple miscarriages (may signify previously affected pregnancies) |
Hyperemesis extending beyond the first trimester | |
Acute fatty liver of pregnancy (AFLP) and hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome association with carrying foetus with long-chain fat oxidation defect | |
Past medical history | Recurrent episodes, especially relating to intercurrent infections (catabolic stress) |
Specific food avoidance, e.g., fructose, protein |
The family history is particularly important searching for evidence for other affected family members such as previous sudden infant deaths, previous miscarriages or known relatives with the same condition or similar presentation. Potential mode of inheritance must be considered and is best facilitated by a three-generation history. Some conditions, such as FAOs, are associated with maternal symptoms in pregnancy including hyperemesis or frank liver dysfunction such as acute fatty liver of pregnancy. Self-imposed dietary restriction is seen in some conditions; especially the avoidance of sweet foods in hereditary fructose intolerance (HFI) and protein in UCDs. HFI will not present prior to weaning as fructose is not present in breast milk or formula milk.
Examination
IMDs associated with obvious dysmorphic features include peroxisomal disorders, CDGs and LSDs ; however, many IMDs have no specific diagnostic features on examination. It is essential to look for extrahepatic manifestations to confirm the multi-organ involvement in some conditions. Developmental concerns are a feature of many IMDs and a full neurological examination should be undertaken to look for neurological signs.
Careful ophthalmic examination is required [2]. Cataracts are nearly always present at birth in galactosaemia but are easily missed in the non-dilated eye due to their transparent (oil drop) appearance. If the diagnosis is missed, the cataract will become more obvious as it matures. Cataracts are also seen in peroxisomal disorders, CTX, Wilson disease and some mitochondrial disorders . A cherry-red spot may be visible due to the accumulation of sphingolipid around the fovea in Niemann–Pick B. Pigmentary retinopathy is a feature of long-chain hydroxy acyl-CoA dehydrogenase (LCHAD) and mitochondrial disorders. Eye movement disorders are a feature of CDG-Ia (squint), NPC (vertical supranuclear gaze palsy), Gaucher disease (horizontal supranuclear palsy) and mitochondrial disorders (external ophthalmoplegia—limited gaze in all directions). Ptosis is found in many conditions including peroxisomal and mitochondrial disorders.
Investigation
Investigations may be divided between the general and the specific. Renal and cardiac assessment including ECG and echocardiography is important to exclude multi-organ involvement. Mitochondrial disorders are characterised by ‘illegitimate associations’, that is, signs and symptoms which may not be obviously linked until one considers the central requirement for energy production. Cardiac involvement may be subtle such as mild compensatory myocardial hypertrophy. Liver imaging may glean further information such as increased reflectivity secondary to fatty steatosis in FAOs and liver biopsy is indicated to help further diagnosis when initial investigations have proven inconclusive. Enzymology and genotyping are often required to secure a diagnosis (see Table 64.3).
Table 64.3
Examples of metabolic investigations and associated pathological findings
Investigation | Condition | Metabolite/finding | Presentation |
|---|---|---|---|
Amino acids | Tyrosinemia type 1 | Raised tyrosine, methionine | C, LF, H |
Urea cycle defects | Raised glutamine, low arginine | LF, E | |
Raised citrulline (citrullinemia) | |||
Raised arginine (arginase deficiency) | |||
Citrin deficiency | Raised citrulline | C, H | |
Galactosaemia | Amino acidemia | C, H, LF | |
Lysinuric protein intolerance | Low lysine, ornithine and arginine | H, E | |
Organic acids | Organic acidemias | Specific organic acids | C, LF, H, E |
Tyrosinemia type 1 | Succinylacetone | C, LF, H | |
Argininosuccinic aciduria (urea cycle defects, UCD) | Argininosuccinic acid | LF, E | |
Ammonia | UCD | LF, E | |
Organic Acidemias | C, LF, H, E | ||
Fat oxidation defects | LF, H, E | ||
Acylcarnitines | Fat oxidation defects | Specific acylcarnitine and often low | LF, H, E |
Free carnitine | |||
Organic acidemias | Specific acylcarnitine and often low | C, LF, H, E | |
Free carnitine | |||
Lactate | Mitochondrial disorders | Raised and exacerbated postprandially | C, LF, E |
Gluconeogenesis | Rapid resolution with treatment | H, E | |
defects | |||
Fat oxidation defects | LF, H, E | ||
GSD I | Elevated on fasting | H | |
GSD III | Elevated postprandially | H | |
CK | Fat oxidation defects | Elevated | LF, H, E |
GSD III | Elevated | H | |
Very long-chain fatty acids (VLCFAs) | Peroxisomal disorders | C, LF, H | |
Gal-1-PUT activity | Galactosaemia | Parental if infant already transfused to confirm carrier levels of activity | C, H, LF |
α1-antitrypsin | α1-antitrypsin deficiency | C, H | |
Lipid profile | Wolman and CESD | Raised lipids | C, H, LF |
Glycogen storage disorders | Raised triglycerides | H | |
Urate | Glycogen storage disorders | Raised | H |
Bile acids | PFIC | Raised normal bile acids | |
Bile acid defects | Specific abnormal bile acids present | ||
Transferrin isoelectric focusing | Congenital disorders of glycosylation | C, H, LF | |
Vacuolated lymphocytes | Lysosomal storage disorders | C, H | |
Glycosaminoglycans (GAGs) and oligosacchraides | Lysosomal storage disorders | C, H | |
Bone marrow aspirate | Lysosomal storage disorders | Storage cells | C, H |
Abdominal X-ray | Wolman | Adrenal calcification | C, H, LF |
Newborn screening offers the opportunity for therapeutic intervention prior to developing clinical symptoms. In the UK, this is limited to a few metabolic conditions including medium-chain acyl-CoA dehydrogenase deficiency (MCADD) which presents with encephalopathy, hypoglycemia and liver dysfunction. Phenylketonuria (PKU), a neurodevelopmental disorder, is screened by detecting elevated phenylalanine; however, a generalised amino acidemia occurs in liver dysfunction and so a positive screen may indicate a liver disorder such as galactosaemia [3]. The clue is phenylalanine to tyrosine ratio which is elevated in PKU due to the block in the conversion of phenylalanine to tyrosine by phenylalanine hydroxylase but normal or decreased in liver disorders. Following an expanded newborn screening pilot, the number of conditions screened is increasing to include isovaleric acidemia and maple syrup urine disease which can both present with decompensation including liver dysfunction in the newborn period. In the other parts of Europe, North America and Australia, many more conditions are covered by newborn screening.
Next-generation sequencing is another technology increasingly used to screen patients to secure a diagnosis. This can either be targeted such as a gene chip, for example, causes of cholestasis or be for a specific group of conditions, for example, GSDs screening all the associated genes simultaneously [4, 5] or screening all coding regions for disease causing mutations (exome sequencing) [6, 7]. This is likely to identify new conditions and broaden the phenotype of known disorders, but is not without risk, ensuring that variations detected are indeed disease causing and responsible for the clinical presentation [8].
Inherited Disorders of Carbohydrate Metabolism
Galactosaemia
Classical galactosaemia (galactose-1-phosphate uridyl transferase deficiency) typically manifests within the first week of life and sometimes within days of starting milk feeds. These feeds introduce galactose that in turn leads to the accumulation of the abnormal and putative toxic metabolite galactose-1-phosphate (see Fig. 64.1). Vomiting and jaundice typically ensue accompanied by hepatomegaly, ascites , deranged liver function, coagulopathy and renal tubulopathy. There is a marked failure to thrive. Careful examination of the eyes reveals oil drop cataracts that are easily missed as the red reflex is present. Classical lentiform cataracts can take many weeks to become apparent and although hypoglycemia is much quoted, it is not a major feature. Neonatal sepsis (typically Escherichia coli) is another mode of presentation and the threshold for antibiotic use in the acute phase should be low. Raised intracranial pressure is also a recognised feature. Occasionally, a more indolent course is followed with symptoms occurring beyond the first month with failure to thrive, renal tubulopathy or the development of cataracts or very rarely developmental delay. However, mild prolongation of the prothrombin time is almost always present.
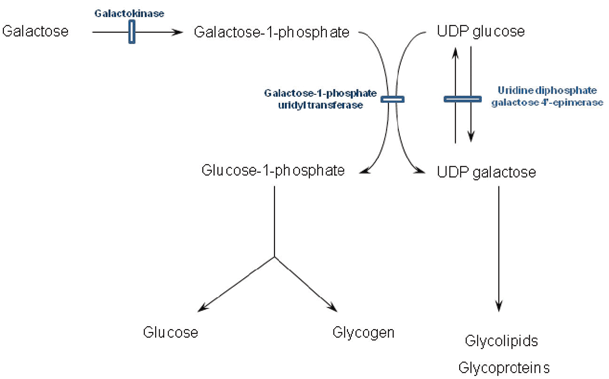
Fig. 64.1
Metabolism of galactose and associated disorders. UDP uridine diphosphate
The incidence in the UK is approximately 1 in 45,000 live births, and the gene is located on chromosome 9 and inherited in a recessive fashion. There is a common mutation Q188R identified in 72 % of cases with classical galactosaemia. Genotype–phenotype correlations are unclear; however, the Duarte variant (N314D) has 50 % residual enzyme activity and is a benign variant [9].
Diagnosis is by measurement of galactose-1-phosphate uridyl transferase activity in erythrocytes. Care must be taken in neonates that have received a red cell transfusion as false negative results can be obtained within 3 months of the transfusion. In this circumstance, galactosaemia can be excluded by testing enzyme activity in the parents and comparing to well-defined heterozygote ranges. Testing urine for the presence of reducing substances is rarely performed as it is neither sensitive nor specific.
Uridine diphosphate galactose 4′-epimerase (UDP-galactose epimerase) deficiency is a rare condition that presents in similar fashion to classical galactosaemia. It should be considered if there is a strong suspicion of galactosaemia when galactose-1-phosphate uridyl transferase activity is normal. It is characterised by elevated galactose-1-phosphate in red cells. Galactokinase deficiency is the third disorder of galactose metabolism, but its phenotype is restricted to ophthalmic symptoms. Galactosaemia screening is available; however, the majority of patients have presented clinically by the time the result would be available and presymptomatic treatment of known sibling cases appears to afford no benefit regarding avoidance of the long-term complications.
If the diagnosis is suspected, galactose must be promptly removed from the diet until the condition has been excluded. Conversion to a soya-based formula, or casein-based protein hydrolysate-based formula (such as Pregestamil) if hepatic impairment is severe, brings rapid improvement. A lifelong dietary restriction of galactose is generally recommended, although practises vary widely and there is much debate regarding the severity of restriction and the necessity of restriction in later life [10]. Many centres will allow a slight relaxation of diet in later life, such as galactose-containing fruit and vegetables while still avoiding sources of lactose, such as dairy products, as this keeps dietary intake well below the level of endogenous galactose production. Calcium intake needs to be regularly assessed to ensure adequate intake, especially in childhood. Long-term complications are not abolished by dietary management, and they include neurodevelopmental problems, osteoporosis and hypergonadotrophic hypergonadism/infertility in females [11]. Careful management of puberty is essential to ensure adequate uterine growth to allow successful pregnancy following ovum donation.
Hereditary Fructose Intolerance
HFI is an autosomal recessive condition due to a deficiency of aldolase B. The key to diagnosis is establishing a clear history of fructose intake prior to the onset of symptoms. Fructose is a monosaccharide that is found in many food sources, notably sucrose (glucose–fructose disaccharide), fruits, vegetables and honey. Sorbitol is an artificial sweetener that is metabolised to fructose. The classical presentation is that of healthy milk-fed infant that develops symptoms when it is first exposed to fructose upon weaning. Gastrointestinal (GI) upset progresses to persistent vomiting, sweating, lethargy and coma if intake continues. These symptoms are accompanied by liver failure and evidence of renal proximal tubulopathy. Hypoglycemia is common but can be masked by glucose administration.
There is a considerable spectrum of disease severity; infants can present with a chronic course of failure to thrive, hepatomegaly and liver impairment. School-age children may present with avoidance of sweet foods and hepatomegaly, with dentition that is unusually free of caries. Patients may develop specific food aversion to avoid sources of fructose that allow them to remain quite well.
Biochemical associations include lactic acidosis, hyperuricemia, marked liver dysfunction and renal tubulopathy. The enzyme deficiency results in accumulation of fructose-1-phosphate, an inhibitor of both glycogenolysis and gluconeogenesis that depletes inorganic phosphate, thus restricting adenosine triphosphate (ATP) production. Cellular depletion of ATP is postulated to be a significant mechanism of hepatocellular toxicity.
There is no rapid test that can exclude HFI. If it is suspected on historical or clinical grounds, then fructose should be immediately excluded. A rapid correction of biochemistry and liver function can be expected over a number of days. Hepatomegaly may take several months to resolve. Direct enzyme assay is restricted to liver tissue, and liver biopsy is likely to be contraindicated in the acute phase. Genotyping of the ALDOB gene is likely to provide a definitive diagnosis.
Treatment is the lifelong restriction of fructose from the diet and is a challenge due to its restrictive nature. Care must be taken to avoid inadvertent consumption of fructose in medicines and processed foods.
Glycogen Storage Disorders
Glycogen is a glucose polymer found primarily in the liver and muscle that is an important source of rapidly accessible stored energy. Hepatic glycogen is ultimately released as free glucose, whereas glycogen in the muscle is utilised directly by the muscle itself. The GSDs are inherited disorders of glycogen breakdown with the exception of GSD type 0 (glycogen synthase deficiency), which is a block in glycogen synthesis causing patients to present with hypoglycemia due to a short fasting tolerance in the absence of hepatomegaly. The clinical manifestation of GSDs reflects the different utilisation of glycogen and can be hepatic (hepatomegaly/hypoglycemia), myopathic or a combination of both. The hepatic forms of GSDs include types I, III, IV, VI and IX (see Table 64.4). Massive hepatomegaly is a feature common to all hepatic GSDs, usually in the absence of splenomegaly although this may be present in a minority of patients in association with hepatic fibrosis and cirrhosis. Hepatomegaly is also a feature of the LSD infantile Pompe disease, also known as GSD type II, although this condition is dominated by hypotonia and cardiomyopathy and is not considered in this chapter.
Table 64.4
Nomenclature of hepatic glycogen storage disorders
Type (Alternate name) | Enzyme | Gene | Inheritance |
|---|---|---|---|
Ia (Von Gierke) | Glucose-6-phosphatase | G6PC | Recessive |
Ib | Glucose-6-phosphate translocase | G6PT1/SLC37A4 | Recessive |
IIIa (Cori/Forbes) | Glycogen debrancher (liver and muscle) | AGL | Recessive |
IIIb (Cori/Forbes) | Glycogen debrancher (liver) | AGL | Recessive |
IV (Andersen) | Glycogen branching enzyme | GBE1 | Recessive |
VI (Hers) | Liver glycogen phosphorylase | PYGL | Recessive |
IXa (XLG) | α-Subunit phosphorylase b kinase (liver) | PHKA2 | X-linked |
IXb | β-Subunit phosphorylase b kinase (liver/muscle) | PHKB | Recessive |
IXc | γ-Subunit phosphorylase b kinase (liver) | PHKG2 | X-linked |
0a | Liver Glycogen synthase | GYS2 | Recessive |
A simplified scheme for liver glycogen metabolism can be seen in Fig. 64.2.
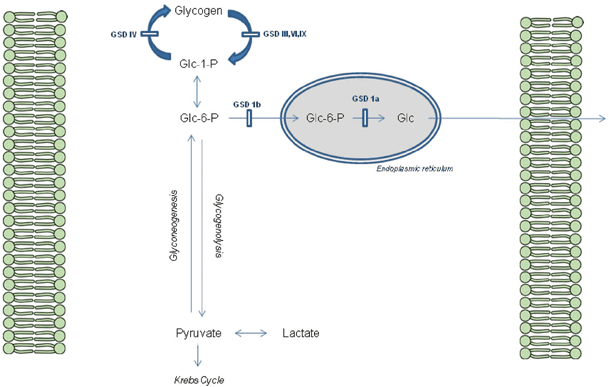
Fig. 64.2
Glycogen metabolism in the Hepatocyte. GSD glycogen storage disorders, Glc glucose, Glc-6-P glucose-6-phosphate, Glc-1-P glucose-1-phosphate
GSD type 1 differs from the other GSDs as it disrupts the release of glucose rather than the breakdown of glycogen itself, effecting both glycogenolysis and gluconeogenesis. Thus, hypoglycemia associated with raised lactate is a hallmark of GSD type 1, in addition to raised urate and triglyceride. It typically presents with hypoglycemia and hepatomegaly in the first 6 months of life when feed frequency is reduced, although a neonatal presentation with hypoglycemia is occasionally seen. Abnormal distribution of fat gives the ‘doll-like’ facies and nephromegaly is a prominent feature. Failure to thrive is common with catch-up growth only occurring with dietary treatment while platelet dysfunction leads to bruising and nose bleeds. GSD type Ib is also associated with neutropenia and abnormal neutrophil function that leads to skin infections, mucosal ulceration and inflammatory bowel disease. Long-term complications are numerous including osteoporosis, gout (hyperuricemia), pancreatitis (hypertriglyceridemia), polycystic ovaries, anemia, Fanconi syndrome and renal impairment. Hepatic adenoma occur in 75 % of patients peri- and postpubertally that have the potential for malignant change [12–15] (see Table 64.5).
Table 64.5
Biochemical hallmarks of hepatic glycogen storage disorders
GSD | Lactate | Urate | Triglycerides | Cholesterol | CK | ALT | Glycogen |
|---|---|---|---|---|---|---|---|
I | ↑ Fasting | ↑ | ↑ ↑ | ↑ | N | ↑ | ↑ Liver |
III | ↑ Postprandial | N | ↑ | ↑ ↑ | ↑ | ↑ ↑ ↑ | ↑ Liver |
↑ Red cell | |||||||
IV | N | N | N | N | N | ↑ ↑ | ↑ Liver |
VI | ↑ Postprandial | N | ↑ ↑ | ↑ | N | ↑ | ↑ Liver |
IX | ↑ Postprandial | N | ↑ ↑ | ↑ | N | ↑ | ↑ Liver |
0 | ↑ Postprandial | N | N | N | N | N | ↓ Liver |
GSD type III is caused by a deficiency of the glycogen debrancher enzyme that results in the accumulation of abnormal glycogen. It may be clinically indistinguishable from type I but fasting tolerance/tendency for hypoglycemia is typically not as severe as type I and nephromegaly is not a feature. Lactate characteristically increases postprandially and creatine kinase is commonly raised. Symptoms gradually improve, particularly around puberty , including spontaneous catch-up of growth and marked reduction in hepatomegaly. The mixed hepatic and muscle form is the commonest (type IIIa). Muscle weakness is not common in childhood and becomes more prominent in adults, although motor milestones may be delayed and activity can be impaired. Cramps at night are common. Long-term outcome in the purely hepatic form (type IIIb) appears excellent. Hepatic adenoma are rare, and reports of cirrhosis and progession to liver failure are infrequent. In type IIIa complications include progressive myopathy and cardiomyopathy although the latter is rarely clinically symptomatic [16, 17].
GSD type IV is an extremely rare condition that classically presents with hepatomegaly and progressive cirrhosis. The deficiency of the branching enzyme produces abnormal glycogen resembling amylopectin. Glucose release from the abnormal glycogen is also impaired and fasting hypoglycemia may occur. Recently, however, the clinical spectrum of this disorder has widened considerably. Different presentations with varying overlap have been reported including foetal akinesia, infantile neuromuscular, cardiomyopathic and adult polyglycosan body disease [18–21]. In neonatal and infantile forms, death within infancy is usual.
Phosphorylase (GSD type VI) and its activator phosphorylase b kinase (GSD IX) are required for the removal of glycosyl molecules from the straight chains of glycogen. GSD type VI presents with pronounced hepatomegaly and a mild tendency to hypoglycemia in early childhood. Growth failure may be marked but catch-up growth occurs spontaneously and normal adult height is achieved. The disorder may be so mild that it remains undiagnosed. Long-term outlook is generally excellent, although hepatic adenomas have rarely been described. GSD type IX has a number of subtypes including three that have a hepatic component (see Table 64.4). Type IXa is by far the most common and has a relatively mild isolated hepatic phenotype similar to type VI. Type IXb is very rare and has a mild mixed liver/muscle phenotype. Type IXc has more severe phenotype often associated with significant hypoglycemia and progression to cirrhosis [22–24].
Confirmation of a suspected diagnosis of GSDs ultimately requires assessment of individual enzyme activity or genetic characterisation. Liver biopsy is not required for the diagnosis but commonly is the mode of diagnosis when patients being investigated for hepatomegaly are found to have significant amounts of glycogen in the liver. Periodic acid–Schiff (PAS) staining reveals cytoplasmic glycogen deposition often accompanied with increased lipid content. The glycogen is of normal structure in GSD types I, VI and IX while types III and IV have abnormal glycogen structure. Type III accumulates glycogen with shortened outer branches and type IV has an amylopectin-like structure [17]. Enzyme analysis requires targeted testing of individual enzymes, with enzymology of type I being limited to liver tissue. With the advent of high-throughput genetic techniques, simultaneous sequencing of all GSD genes is now available and is superseding enzyme assays, although the enzyme assay remains a valuable tool to evaluate genetic changes whose pathogenicity is uncertain.
Treatment for GSD type I includes strict dietary management. Hypoglycemic episodes are avoided by instituting frequent high-carbohydrate feeds during the day and continuous overnight tube feeds. This may be supplemented with uncooked or modified cornstarch to prolong normoglycemia [25], acting as a slow release form of glucose. Prophylactic trimethoprim is beneficial in GSD Ib for oral ulceration and granulocyte colony stimulating factor (GCSF) is reserved for resistant cases or those with recurrent infection. Regular assessment is required to monitor for long-term complications such as renal disease, hepatic adenoma/malignant transformation, osteopenia, hyperuricemia and hyperlipidemia [15]. Dietary management in type III depends on the severity [17]. Patients with early-onset hypoglycemia are managed as type I. A high protein intake has been advocated as gluconeogenesis is intact [26]. Dietary treatment in types IV, VI and IX is often unnecessary, but there is evidence that dietary management may improve fibrosis and long-term outcome in type IX [27].
Liver transplant is rarely considered but has been used in cases of hepatic failure, adenoma or malignancy or in cases refractory to dietary manipulation in patient with GSD types I and III [28, 29]. The only significant therapeutic intervention for type IV is liver transplant, although the extrahepatic progression of neuromuscular and cardiac disease post transplant has been documented in a number of patients [30].
Fanconi–Bickel Syndrome
The Fanconi–Bickel syndrome, previously classified as type GSD XI, presents with marked hepatomegaly secondary to glycogen storage and fasting hypoglycemia. Other features include postprandial hyperglycemia, hypergalactosaemia and renal Fanconi syndrome. The primary defect is a deficiency of the glucose 2 (Glut2) transporter important in the uptake and release of glucose from the liver. Glut2 is also expressed in pancreas, gut and kidney. Defects impair glucose sensing within islet β-cells compounding hyperglycemia in the fed state, due to decreased liver uptake, by blunting the insulin response. Hypoglycemia, in the fasting state, is secondary to impaired glucose release from the liver. Increased intrahepatic glucose inhibits glycogen degradation facilitating storage, and glycogen deposition may be noted on liver biopsy. Diagnosis relies on recognition of abnormal glucose homeostasis and renal tubulopathy while transaminitis, hyperlipidemia and hyperuricamia may be present. Treatment focuses on dietary support of glucose homoestasis with regular feed and also active replacement of renal losses.
Fructose-1,6-Bisphosphatase Deficiency
Fructose-1,6-bisphosphatase (FDP) deficiency is a defect in gluconeogenesis, the pathway that generates endogenous glucose and is a crucial mechanism to maintain glucose homeostasis when dietary glucose is depleted. It is also required for the metabolism of exogenous fructose. The classical features of FDP are hypoglycemia, lactic acidosis and prominent hepatomegaly. Approximately, 50 % of cases present within the first 4 days of life and the majority by 6 months of age. Acute acidosis causes hyperventilation and irritability that progress to coma, apnoea and cardiac arrest. Acute hepatomegaly is commonly found, and a urinary ketosis can be evident (an unusual finding in neonates). Diagnosis relies on clinical suspicion that is confirmed by enzyme analysis of FDP in leukocytes (or liver) or genetic analysis of the FBP1 gene. It is an autosomal recessive condition and a family history of neonatal death/acidosis is not uncommon.
Treatment of the acute presentation involves vigorous supplementation of glucose to inhibit gluconeogenesis. The acidosis and hepatomegaly generally respond promptly to this therapy. The acidosis may be severe enough to warrant use of bicarbonate to correct acid–base balance. If the sequelae of acute hypoglycemia and acidosis are avoided, prognosis is excellent. Mild hepatomegaly may persist during infancy but is not associated with signs of liver disease. Chronic treatment involves strict avoidance of fasting and adequate supply of energy during intercurrent illness. Dietary sources containing significant amounts of fructose (includes sucrose/sorbitol) should be avoided but fructose does not have to be rigorously excluded as in HFI.
Transaldolase Deficiency
Transaldolase deficiency is a very rare single enzyme defect of the pentose phosphate pathway. There seems to be a wide phenotype but the few cases described presented uniformly in the neonatal period with hepatosplenomegaly and liver impairment [31]. Associated features included dysmorphism (including cutis laxa), cardiac anomalies, oedema, renal abnormalities, hemolytic anemia and thrombocytopenia.
Prognosis is poor with developmental delay and commonly death within the first year of life associated with liver failure. Diagnosis relies on clinical suspicion followed by analysis of urine polyols (showing amongst others raised sedoheptulose-7P). Confirmation of diagnosis can be achieved by enzyme assay in lymphocytes, erthyrocytes, fibroblasts or liver tissue and by analysis of the TALDO1 gene.
Congential Disorders of Glycosylation
The CDGs are a large group of disorders that present a considerable diagnostic challenge. Over the past decade, their number has rapidly expanded to nearly 70 discrete conditions. Individually, they are rare disorders with a very broad phenotype whose biochemical diagnosis relies on complex and often non-specific methods (i.e. transferrin isoelectric focusing). Glycosylation is the addition and modification of complex carbohydrate molecules (glycans) to proteins and lipids. The majority of extracellular, membrane bound and some intracellular proteins are glycosylated and the glycans perform a wide variety of functions from structural roles to cell–cell signalling. The process of glycosylation is a highly complex intracellular mechanism, the understanding of which is constantly being updated as new disorders are discovered [32].
The majority of CDGs are multisystem disorders. Developmental, skeletal and neurological problems dominate many of these conditions while hepatic involvement is not uncommon. Typical of this is by far the most common CDG, phospho-mannomutase 2 deficiency (PMM2-CDG). Even within this single condition, the phenotypic spectrum is very wide, but patients classically present in the neonatal period with dysmorphism, abnormal fat pads, inverted nipples and hepatomegaly. Significant development delay becomes evident with hypotonia, ataxia and failure to thrive. Transaminitis is typical and liver histology is characterised by steatosis and fibrosis with myelin-like lysosomal inclusions within hepatocytes on electron microscopy
Phosphomannose isomerase deficiency (MPI-CDG) is a rare CDG that presents primarily with hepatic and GI phenotype, but is remarkable as the sole CDG with an effect treatment. Typical presentation is within the first year to life with vomiting, abdominal distension, protein losing enteropathy, GI bleeding and liver dysfunction. Hypoglycemia may also be apparent. Dysmorphic features and a significant neurological phenotype are not associated with this condition. Transferrin isoelectric focusing is abnormal (see below) and diagnosis can be confirmed by enzymology in leukocytes or by gene sequencing ( MPI gene). Treatment is by dietary supplementation of mannose [33].
Diagnostic testing for CDGs starts by analysing the glycan structures on a well-characterised glycoprotein such as transferrin (transferring isoelectric focusing). An abnormal pattern can then undergo further biochemical testing, or if the clinical picture permits, a specific genetic diagnosis can be sort. It should be noted that other liver pathology can result in an abnormal transferrin isoelectric focusing pattern, namely alcoholism, classical galactosaemia and HFI. When liver disease is present as part of an undefined systemic disorder, a congenital disorder of glycosylation should be considered [34].
Inherited Disorders of Protein Metabolism
Tyrosinemia Type 1
The metabolic defect responsible for tyrosinemia type 1, fumarylacetoacetase deficiency, arises late in the catabolic pathway of tyrosine (see Fig. 64.3). The immediate precursors to the block fumarylacetoacetate and maleylacetoacetate damage the liver and kidney, with their reduced derivatives including succinylacetone. The early onset form presents with liver failure, coagulopathy, jaundice and ascites in the first 6 months of life. Hypoglycemia may result from liver dysfunction or hyperinsulinism secondary to islet cell hyperplasia [35]. Milder forms may present with hypophosphatemic rickets secondary to a renal Fanconi syndrome. Occasionally, children present with neurological crises which resemble acute porphyria with abdominal pain , hypertension, peripheral neuropathy and muscle weakness.
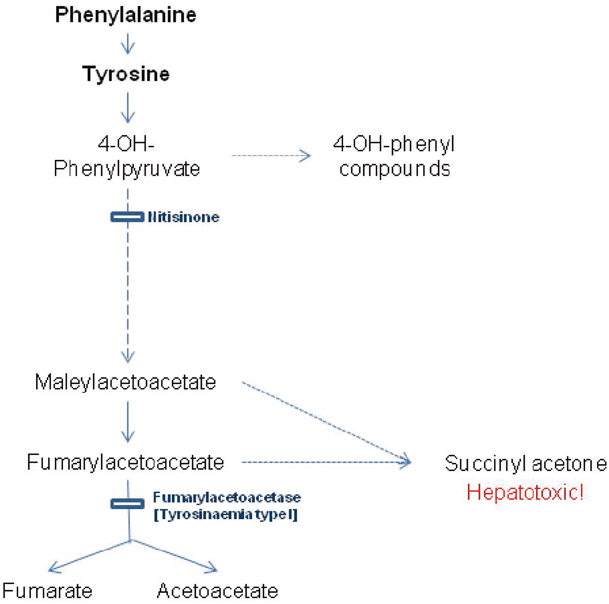
Fig. 64.3
Diagram showing biochemical pathway of tyrosine catabolism, defective step causing tyrosinemia type I and the action of the substrate reduction therapy nitisinone
Liver function reveals mild elevation of bilirubin, transaminemia, elevated alakaline phosphatase and deranged coagulation. Tyrosine, phenylalanine and methionine are raised on plasma amino acids in acutely ill patients. The presence of succinylacetone on urinary organic acids is pathognomonic, and the diagnosis is confirmed on genotyping although enzymology in lymphocytes or fibroblasts can be performed. α-Fetoprotein is raised acutely and falls with treatment. Urinary ∆-aminolevulinic acid is raised. Biochemical evidence of a renal Fanconi syndrome is common. Hypertrophic cardiomyopathy is described.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


