8. DIGESTION AND ABSORPTION
After studying this chapter you should be able to:
1. Explain the mechanisms of digestion and absorption of complex nutrients, vitamins and minerals.
2. Understand the consequences of malabsorption in intestinal diseases.
Introduction
Most digestion and absorption occurs in the small intestine. The transport of water, monovalent ions and drugs was discussed in Chapter 7. In this chapter, the digestion of complex nutrients, and the absorption of the products of digestion, as well as the absorption of vitamins and minerals will be considered.
The consequences for nutrition, of disease of the small intestine will also be addressed in this chapter. As absorption of different nutrients can occur in different regions of the gastrointestinal tract, the regions affected by the disease process determine which nutrients will be poorly absorbed. Coeliac disease (Case 8.1, Case 8.1 and Case 8.1), which usually affects the proximal small intestine, and Crohn’s ileitis (Case 8.2, Case 8.2 and Case 8.2), which usually affects the terminal ileum, will be used to illustrate some of the general principles of absorption, as well as the specific problems encountered as a consequence of malabsorption of the nutrients that are normally absorbed in the affected regions.
Case 8.1
A 25-year-old woman visited her doctor and complained of diarrhoea and flatulence. She also said she had recently lost a considerable amount of weight, and she felt weak and exhausted most of the time. She also suffered from back pain. Upon questioning she said her faeces were bulky, greasy and foul-smelling.
She recalled that she had had persistent diarrhoea throughout childhood but the symptoms had disappeared during adolescence. She was referred to a gastroenterologist. The consultant arranged for blood and faecal analyses. The faecal tests confirmed the presence of steatorrhoea. The blood tests indicated that she had iron-deficiency anaemia, folate-deficiency and Ca2+ deficiency. Her blood electrolyte concentrations and prothrombin clotting time were within the normal range. The consultant suspected coeliac disease and arranged for an endoscopy (telescopic visualization of the duodenum) to be performed. A biopsy of the mucosa, taken at the examination, showed flattening of the villi and excessive plasma cells in the submucosa. A further blood test, to measure the concentrations of transglutaminase antibodies was performed and this showed a high titre of the antibodies. In view of these findings the consultant told the patient to exclude wheat, rye and barley flours (but not oat flour) from her diet, and to try to ensure that it was nutritionally balanced. She was prescribed iron, folate and vitamin D supplements. This diet was not easy to follow as so many food products contain the flours, but after a few weeks the patient had vastly improved. She had gained weight and was no longer feeling constantly tired.
After reading this case history we can address the following questions:
• What is the basic defect in this condition?
• If the duodenum and proximal jejunum were the regions of the small intestine involved, which nutrients are likely to be malabsorbed? Why was iron-deficiency anaemia present? Why was the patient’s clotting time measured? Is milk intolerance likely to be a complication in this condition? Why was her blood investigated for transglutaminase antibodies?
• Why was steatorrhoea present in this patient?
• What problems result from deficiencies of these nutrients?
• How would a defect in CCK and secretin release from the duodenum affect the functioning of the digestive system?
• What are the likely causes of diarrhoea in coeliac disease?
• Why were the patient’s blood electrolyte concentrations measured?
Case 8.2
A 17-year-old young man complained to his general practitioner that he had been suffering from abdominal pain, diarrhoea, weight loss and feelings of lassitude. The doctor examined him and found that his abdomen was distended. He ascertained that the pain was in the central and right lower quadrant. He suspected acute appendicitis, and the patient was admitted to hospital. An abdominal operation was arranged. The surgeon observed that the appendix appeared normal. However, a short length of the terminal ileum was reddened, thickened and oedematous. These features indicate Crohn’s disease of the terminal ileum. No further surgery was performed. Following the operation, blood and faecal samples were obtained for analyses. The patient was allowed a few days to recuperate and was then sent home. He was prescribed codeine for the pain, and diphenoxylate (Lomotil) for the diarrhoea, and he was started on oral steroids to reduce the inflammation. He was advised to keep to a nutritious diet. The acute symptoms gradually settled, but he suffered several relapses over the next few years. He required iron supplements and intramuscular injections of vitamin B12. He developed steatorrhoea.
The patient eventually suffered an intestinal obstruction. He was maintained on intravenous parenteral nutrition for 2 weeks and during this time the symptoms diminished, but they returned when he resumed normal nutrition. An emergency operation was then performed to remove the affected part of the ileum (which was causing the obstruction). Following the operation he made a good recovery. He resumed a normal diet and regained the weight he had lost. Later in life he developed symptomatic gallstone disease that required the removal of his gall bladder (cholecystectomy).
Upon consideration of the details of this case we can address the following questions:
• What is the basic defect in Crohn’s disease and what causes it? Which parts of the gastrointestinal tract can be affected in this disease?
• How is the condition diagnosed? Which blood and faecal analyses would have assisted the diagnosis?
• What could account for the symptoms of weight loss and lassitude? Which nutrients are poorly absorbed in Crohn’s disease? Why were diarrhoea and steatorrhoea present? Why was the patient started on iron supplements? Why was he given intramuscular vitamin B12?
• Why was the patient maintained on parenteral nutrition for a while? What was the likely composition of the intravenous fluid used?
• What are the likely causes of the intestinal obstruction?
• What could be the cause of the gallstone disease that developed later in life in this patient?
Absorption
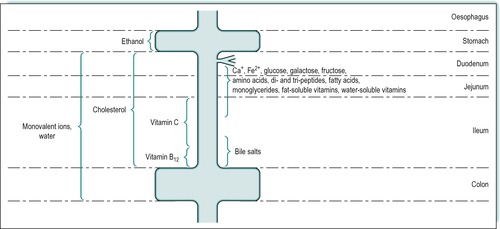
Most nutrients are absorbed at a slow rate by passive diffusion throughout the small intestine. However, many important nutrients are absorbed at a faster rate by processes which involve saturatable mechanisms (see Ch. 1). The proximal small intestine, i.e. the duodenum and jejunum, is the location of most of these special mechanisms, as most substances are absorbed predominantly in those regions. Figure 8.1 shows the approximate sites of absorption of many important nutrients. The important divalent cations, Ca2+ and Fe2+, are absorbed mainly in the duodenum and jejunum. Hexoses, including glucose, galactose and fructose, are also absorbed in the duodenum and jejunum, as are amino acids, small (di- and tri-) peptides and some water-soluble vitamins. Fatty acids, monoacylglycerols and fat-soluble vitamins are also absorbed in the duodenum and jejunum. Cholesterol is absorbed throughout the small intestine. Vitamin C is absorbed in the proximal ileum. Vitamin B12 and bile salts are absorbed predominantly in the terminal ileum. Water and monovalent ions are absorbed throughout the small and large intestines. The defects in absorption seen in coeliac disease and Crohn’s disease which affect different regions of the small intestine, are described in Case 8.1 and Case 8.2.
Case 8.1
Defect and treatment
Coeliac disease is common in Northern Europe with an incidence of possibly 1 in 250. (It is rare in Africa.) Genetic and environmental factors are involved.
Defect
The disease is due to an abnormal reaction to gluten, a constituent of wheat flour. Gluten damages the enterocytes, causing atrophy of the villi, and malabsorption. The duodenum and proximal jejunum are usually more severely affected than the ileum. The damage is due to an abnormal immune response to gliadins (especially α-gliadin), components of gluten. It is a T-cell-mediated disease. Antibodies to the enzyme transglutaminase, which is released in tissues during inflammation, are present in 98% of affected individuals. The mechanisms involved in the damage have not yet been fully characterized but there is evidence that deamidation of gliadin by transglutaminase generates a recognition site for CD4+ T lymphocytes; the locally activated lymphocytes trigger production of cytokines which then cause the damage. In addition transglutaminase antibodies have been shown to affect the differentiation of epithelial cells, possibly by interfering with the action of the enzyme.
Diagnosis
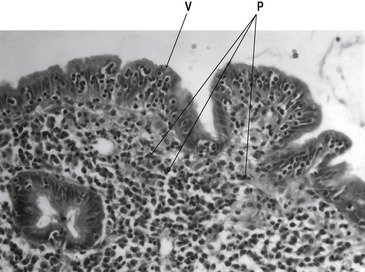
Determination of the serum concentration of transglutaminase antibodies is a useful tool in the diagnosis of coeliac disease. The diagnosis of coeliac disease also requires a biopsy, usually of the duodenum, which would show flattened villi, crypt hyperplasia, infiltration with lymphocytes and plasma cells and reduced cell differentiation. Figure 8.2 shows a section through the jejunal mucosa in a patient with coeliac disease. The cells cannot be replaced quickly enough by stem cell division in the crypts, and many of the cells present are immature and therefore do not absorb nutrients effectively.
Treatment
Treatment of coeliac disease involves the individual following a gluten-free nutritious diet. The gliadin peptides that cause the abnormal reactions are present in wheat, rye and barley but not oats. The latter is therefore safe for individuals with coeliac disease.
Case 8.2
Defect, diagnosis and treatment
Crohn’s disease is most commonly diagnosed in young adults. The highest frequency is seen in Caucasians, and in the Western world. In the UK, it affects approximately 50/100 000 of the population. The cause is unknown, but it is probably multifactorial. As it is an inflammatory disease, infectious organisms, including the measles virus and Mycobacterium pseudotuberculosis, have been variously implicated. Autoimmunity has also been suspected because there is an association with known autoimmune conditions such as arthritis and eczema. Inherited factors are also implicated because of the high concordance in monozygotic twins, and in families. Crohn’s disease in children can lead to growth retardation and delayed sexual development, probably because of poor nutrition.
Defect
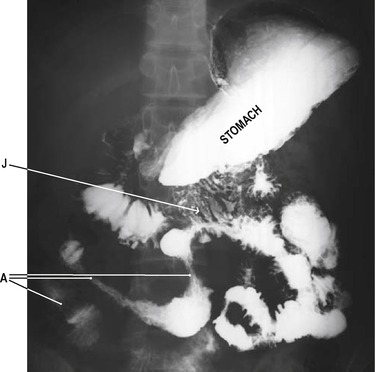
Crohn’s disease is an inflammatory disorder that can affect any region of the gastrointestinal tract, from the mouth to the anus. However, the terminal ileum is the commonest site to be affected (Crohn’s ileitis, Fig. 8.3). In the mouth, aphthous ulcers of the buccal mucosa and tongue are seen. At the anus, skin tags, fissures and fistulae may be present. The disease is characterized by remissions and relapses. Macroscopically, the intestines appear red and swollen. Normal areas of tissue are usually present between the damaged areas. Because the inflammatory process involves all layers of the intestinal wall, diseased segments become grossly thickened. This can give rise to obstruction. As the inflammation resolves, areas of secondary scarring (fibrosis) in the wall of the bowel also constitute obstructive regions. The mucous membrane appears ‘cobble-stoned’ due to longitudinal fissures and transverse oedematous folds. Aggregations of inflammatory cell infiltrates may be present, and the mesenteric lymph nodes may be enlarged due to reactive hyperplasia. Granulomas (aggregates of epithelial macrophages surrounded by a cuff of lymphocytes) may be present in the lymph nodes and the bowel wall.
The formation of enteric fistulae is a further feature of the disease. Fistulae may extend between different loops of the bowel (enteroenteric fistulae), or between the bowel and the skin (enterocutaneous fistulae, especially in the perianal areas). Gastrointestinal bleeding can also occur. This is usually mild but, because it is chronic, it can lead to iron-deficiency anaemia. There is also an increased incidence of malignant neoplasms in patients with Crohn’s disease, especially in the small and large bowel. This may be related to the long-term damage to the bowel mucosa.
Diagnosis
The diagnosis of Crohn’s disease is difficult because all the salient features are seen in other disorders. A combination of histology, endoscopy and radiology is usually employed. Evidence from blood analyses, including leukocytosis, elevated sedimentation rate, and thrombocytosis, is indicative of an active inflammatory process. Radiological evidence of the sites of involvement, and the chronic remitting course of the patient’s illness, also assist the diagnosis. However, a definitive diagnosis requires histological assessment of the bowel and identification of granulomata.
Treatment
The management of patients with this disease includes:
• Treatment of the symptoms (diarrhoea, pain)
• Treatment with anti-inflammatory and immunosuppressive agents
• Surgical treatment where there are complications such as luminal obstruction
• Management of the patient’s nutritional status, including supplementation of the diet
• Parenteral nutrition when necessary.
Total parenteral nutrition
Intravenous feeding can be used in extreme cases to rest the bowel and allow healing. The fluid administered would contain amino acids, glucose and lipid in amounts sufficient to meet the protein and energy needs of the individual, and electrolytes, vitamins and minerals in amounts sufficient to meet the estimated daily requirements. In patients with Crohn’s disease total parenteral nutrition can lead to positive nitrogen balance, weight gain and temporary remission of symptoms.
Absorption of important nutrients
Carbohydrates
The average daily intake of carbohydrate in the human adult is probably between 250g and 800g/day. The useful carbohydrate in the food is largely vegetable starch in potatoes, bread, pasta and rice, and to a lesser extent glycogen (animal starch), in meat and liver. These polysaccharides are composed entirely of D-glucose subunits linked together mainly by α-1,4 glycosidic linkages. In the human, over 90% of the starch in the diet is digested and absorbed. The remainder passes into the colon where it may be utilized by colonic bacteria.
Cellulose is another polysaccharide, which is composed of glucose subunits, present in the diet. However, it is not digestible in humans and other non-ruminants because the subunits are linked by β-1,4 glycosidic bonds that cannot be hydrolysed by the enzymes in the digestive tract. Therefore it passes into the colon. Nevertheless, cellulose is an important source of dietary fibre, providing bulk that stimulates intestinal motility, and prevents constipation (see Ch. 10). In ruminants, cellulose is degraded by bacterial cellulases that hydrolyse the β-1,4 glycosidic linkages to produce D-glucose, which is absorbed.
There are appreciable amounts of disaccharides, including sucrose (table sugar), lactose (milk sugar) and maltose (malt sugar), in Western diets. The only free monosaccharide likely to be present in the diet is glucose, which is added to ‘high energy’ drinks and foods.
Dietary carbohydrate is utilized to provide energy for muscular and secretory activity and other metabolic functions. It is not ‘essential’ as a source of energy as calories can also be provided by fat and protein. However, a few carbohydrate substances, such as inositol, are ‘essential’ vitamin components of the diet, as they either cannot be synthesized in the body, or cannot be synthesized at a rate which is rapid enough to meet the body’s requirements.
Structure of starch and glycogen
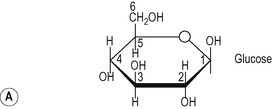
The molecular weight of vegetable starch ranges from a few thousand to 500 000. It consists of two components:
1. Amylose in which the glucose subunits are linked together in straight unbranched chains via α-1,4 glycosidic linkages (Fig. 8.4A).
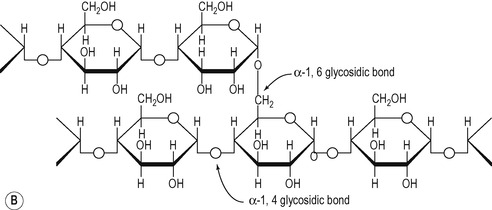
2. Amylopectin which consists of branched chains, with the branches occurring at approximately every 30th glucose residue. α-1,4 linkages are present within the chains and α-1,6 linkages occur at the branch points (Fig. 8.4B).
Digestion of carbohydrate
In the gastrointestinal tract there are several enzymes that degrade starch and glycogen. These include the α-amylases secreted by the salivary glands and the pancreas, and isomaltase and glucoamylase, which are integral components of the intestinal absorptive cell membranes. Maltose, sucrose and lactose can also be degraded to their component monosaccharides by enzymes situated in the brush border of the upper small intestine (see below).
α-Amylases
α-Amylases split the α-1,4 glycosidic linkages in amylose to yield maltose and glucose, but they do not act on maltose, a disaccharide composed of two glucose subunits linked by an α-1,4 linkage. In theory α-amylase will ultimately degrade a solution of amylose to maltose, and glucose which can be released from the ends of the chains (Fig. 8.5). Intermediate oligosaccharides (dextrins) are formed in the process. α-Amylases also attack amylopectin and glycogen at their α-1,4 linkages. Intermediate unbranched oligosaccharides and branched oligosaccharides (α-limit dextrins) are formed. Thus a mixture of products is produced (Fig. 8.5).
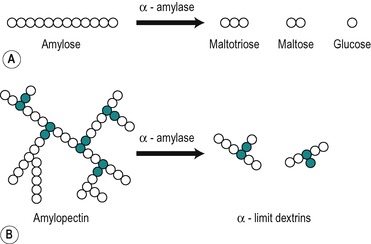
Salivary amylase starts the digestion of starch. It continues to act for up to half an hour in the interior of the food bolus after it has arrived in the stomach. It is eventually inactivated at the low pH produced by the gastric acid when it penetrates the food bolus. It can digest up to 50% of the starch present in food. Pancreatic juice that contains a second α-amylase is released into the duodenum when a meal is present in the digestive tract. Pancreatic amylase continues the digestion of starch and glycogen in the small intestine. It is produced in larger amounts than salivary amylase. The α-amylases from the two sources have similar catalytic properties, despite having different amino acid sequences. They both require Cl− for optimum activity and both act at neutral or slightly alkaline pH values.
Role of brush border enzymes
Intestinal isomaltase (α-1,6 glycosidase) splits α-1,6 linkages in the branched poly- and oligosaccharides produced by amylase action in the small intestine. The combined action of α-amylase and α-1,6 glycosidase can degrade amylopectin and glycogen to a mixture of maltose and glucose, but other enzymes in the brush border speed up and complete the process of starch digestion. These are glucoamylase, which degrades small unbranched oligosaccharides, and maltase and isomaltase, which degrade maltose and isomaltose, respectively. All of these enzymes have access to the mixture of polysaccharides, oligosaccharides and disaccharides in the chyme during a meal.
The enzymes available to digest disaccharides are:
• Maltase that degrades maltose to glucose
• Sucrase that degrades sucrose to glucose and fructose
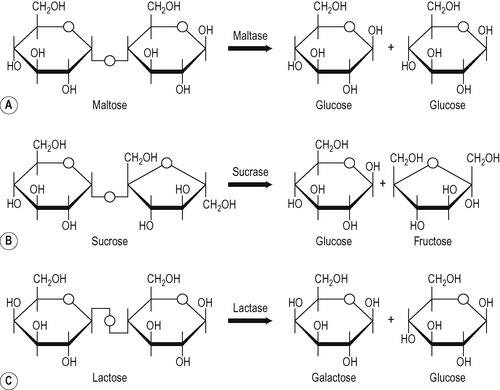
Sucrase and isomaltase are synthesized as a single polypeptide chain inside the cell, and this is inserted intact into the plasma membrane. Pancreatic protease cleaves the polypeptide chain at a site between the active centres of the two enzyme moieties, but they remain non-covalently associated in the membrane. When sucrase is incorporated into artificial membranes, it binds sucrose at one face and releases glucose and fructose on the other side of the membrane. This has led to speculation that it is both a hydrolytic enzyme and a transport molecule. The other brush border enzymes do not appear to behave in this way. These disaccharidases are all present in the brush border of the enterocyte, and it seems that the digestion of disaccharides and small oligosaccharides actually occurs in the membrane itself. This has been inferred from the fact that after administration of a solution of a disaccharide to an animal, very little free glucose can be detected in the lumen, yet the disaccharide molecules are too large to diffuse through the pores in the membrane, and disaccharide molecules cannot be detected in the blood. It seems likely that the brush border enzymes occupy positions in the membrane that are adjacent to the hexose carriers. The release of monosaccharides occurs on the surface of the membrane and these are then transferred to the adjacent carrier molecule for transport into the cell (Fig. 8.7). Inherited disorders of brush border enzymes are described in Box 8.1. These defects result in carbohydrate malabsorption and diarrhoea.
Monosaccharide absorption
The most abundant monosaccharides in dietary carbohydrate are the hexoses, D-glucose, D-galactose, and D-fructose, the products of digestion of starch, sucrose and lactose. Both L-hexoses and D-hexoses are absorbed slowly by passive diffusion in the gastrointestinal tract. However, the plasma membrane of cells is relatively impermeable to polar molecules such as monosaccharides and the transport of these sugars into the enterocyte by passive diffusion is therefore slow. Glucose, galactose and fructose are absorbed by saturatable mechanisms, mainly in the duodenum and jejunum. This is accomplished by membrane-associated transporters located in the brush border and basolateral membranes of the mature enterocytes. These bind the sugars and transfer them across the cell membranes and deliver them to the interstitial fluid in the lateral spaces, from where they are taken up into the adjacent capillaries to enter the portal blood.
Pentoses are smaller than hexoses but they are absorbed at a slower rate than glucose, galactose and fructose, indicating that they are probably absorbed by passive diffusion.
Hexose transporters
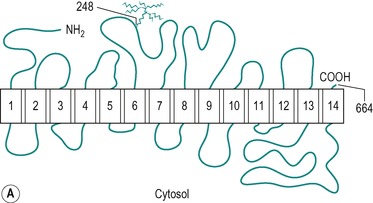
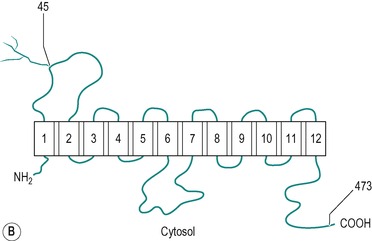
There are two types of hexose transporter in mammalian cells; Na+/glucose co-transporters which are involved in the secondary active transport of glucose, and Na+-independent facilitative hexose transporters. Two forms of the Na+-dependent transporter have been identified. These are SGLT1 and SGLT2, but only SGLT1 is present in the small intestine. There are at least five functional isoforms of facilitative transporter (GLUT1, GLUT2, GLUT3, GLUT4 and GLUT5). All mammalian cells express at least one of these transporters. The most studied is GLUT4, an insulin-sensitive glucose transporter present in muscle and adipose tissue (see Ch. 9). GLUT1, GLUT2 and GLUT5 are all present in the enterocyte. Figure 8.8 shows the structures of the SGLT1 and GLUT1 transporters, and their conformations in the membrane. The two molecules exhibit many similar features. The specificity of the transporters is shown in Table 8.1. An inherited disorder where the SGLT1 is absent from the brush border has been described (see Box 8.1).
| Transporter | Glucose | Galactose | Fructose |
|---|---|---|---|
| SGLT1 | + | + | − |
| GLUT1 | + | + | − |
| GLUT2 | + | + | + |
| GLUT5 | − | − | + |
Box 8.1
Brush border membrane disease is a collective term for conditions in which there is an absence of, or a marked decrease in the activity of a functional brush border membrane protein. These diseases are usually genetic. Sufferers may also exhibit disorders of reabsorption in the kidney proximal tubules. Several brush border diseases exist where the defect results in malabsorption of a specific carbohydrate. Intolerance of that carbohydrate in the diet develops. The unabsorbed sugar passes into the colon where some of it is fermented by bacteria. The remaining unaltered sugar and its bacterial fermentation products cause osmotic diarrhoea (see Ch. 7). The clinical symptoms of carbohydrate malabsorption are abdominal distension, gassiness, borborygmi, nausea, cramping, pain and diarrhoea. In some of these conditions, there is also a high incidence of ulceration of the mouth. Carbohydrate malabsorption is diagnosed by an oral tolerance test that involves administration of the suspected sugar by mouth, and measurement of the sugar in the blood and faeces. If the individual is intolerant of the sugar, it appears in the faeces, but it (or its normal digestion products) cannot be detected in the blood. Confirmation is by examination of a jejunal biopsy to see if the appropriate enzyme or carrier protein is absent from the mucosa.
Brush border diseases that have been characterized include:
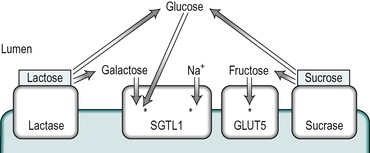
• Lactase deficiency. This is a common brush border disorder that is usually expressed in adolescence or early adulthood. The enzyme, lactase, is induced by its substrate, lactose, in individuals who consume milk. There is generally a high incidence of lactase deficiency in populations such as Mediterranean and Oriental races that do not normally consume milk after childhood, with the result that the enzyme is not present in their intestines. If milk is ingested in these individuals they experience the symptoms of lactose intolerance. A rare congenital form of lactase deficiency exists; feeding infants with this condition with normal milk causes diarrhoea. The problem can be overcome by feeding artificial milk in which lactose has been replaced by sucrose or fructose. There is a rise in H2 elimination in the breath of affected individuals: the result of the metabolism of the unabsorbed lactose by bacteria in the colon.
• Sucrase–isomaltase deficiency. Individuals with this inherited brush border enzyme disease are intolerant of sucrose and isomaltose.
• Glucose/galactose malabsorption. In this condition, there is a genetic defect in the Na+-dependent glucose/galactose transporter, SGLT1. Ingestion of glucose or galactose produces the symptoms of brush-border disease. Treatment involves omitting these sugars, as well as lactose (which is degraded to glucose and galactose) from the diet. Fructose is absorbed normally via the GLUT5 transporter.
Hexose transport
The uptake of glucose across the enterocyte plasma membrane involves the binding of glucose to the Na+/glucose cotransporter SGLT1, as described in Chapter 7. SGLT1 is present only in mature enterocytes in the upper regions of the villi. Galactose also binds to this carrier, but fructose does not. Glucose and galactose transport into the epithelial cell is via secondary active transport. The energy required is derived from the coupling of sugar transport to the transport of Na+ down the concentration and electrical gradients from the lumen into the cell. Both Na+ ions and the sugar are transported into the cell on the SGLT1 transporter. This represents a major route for the uptake of both the sugar and Na+ into the enterocyte. Thus the uptake of glucose is stimulated by the presence of Na+ in the intestinal chyme. The affinity of the carrier for glucose increases as the luminal Na+ concentration increases. The Km of SGLT1 in the presence of Na+ is less than 0.5mM, but in its absence it is greater than 10mM. The absorption of glucose is illustrated schematically in Figure 8.9. Each carrier molecule binds two Na+ ions and one glucose molecule. The glucose concentration in the cell may be higher than that in the lumen but the coupling of the transport of glucose with that of Na+, enables glucose to be transported into the cell against a concentration gradient.
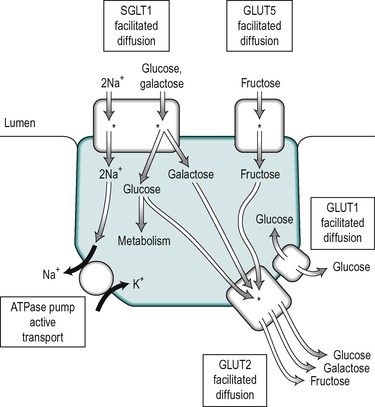
Fructose does not bind to SGLT1 in the brush border. It is transported into the enterocyte, down its concentration gradient by GLUT5 (which does not transport glucose, Fig. 8.9). The separate pathways for glucose and fructose transport into the enterocyte can be inferred from the fact that normal fructose absorption is present in patients with inherited glucose-galactose malabsorption (see Box 8.1), and this provides the rationale for the treatment of this condition with fructose. GLUT5 is present only in mature enterocytes on the tips and sides of the villi in the jejunum.
Some glucose is utilized by the cell for its energy requirements. The transport of the remaining glucose, galactose and fructose across the basolateral membrane is accomplished by the GLUT2 transporter, which has a low-affinity for glucose (Km 23mM). The presence of this low-affinity transporter allows the rate of glucose transport through the basolateral membrane to increase in proportion to the glucose concentration, which varies from 5mM (the normal value) to 20mM. GLUT1 is also present in enterocytes but its function is unclear. It is a high-affinity transporter that functions close to the Vmax even at normal blood concentrations. It may participate in the release of glucose at the basolateral border. However, in other tissues, such as the kidney tubules, where it is present in the basolateral membranes its function appears to be to provide the cells with a source of metabolic energy derived from the blood. Its function in the enterocyte may therefore be to provide glucose from the blood for metabolism during periods of fasting when it is not being absorbed from the intestinal lumen. The locations of the different transporters in the enterocyte are illustrated in Figure 8.9.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


