Fig. 3.1
Common anatomical variants of EA/TEF anomalies. a EA with distal TEF, b isolated EA with no TEF, c H-type TEF, d proximal and distal TEF, e EA with proximal TEF. (Reprinted with permission from [4], Fig. 2.1. EA esophageal atresia, TEF trache-esophageal fistula)
Type of lesion | Frequency (%) |
|---|---|
EA and distal TEF | 88.8 |
Isolated EA | 7.3 |
H-type fistula | 4.2 |
Distal and proximal TEF | 2.8 |
Proximal TEF | 1.1 |
Group | Clinical features | Survival (%) |
|---|---|---|
I | BW ≥ 1500 g with no major CHD | 97 |
II | BW < 1500 g or major CHD | 59 |
III | BW < 1500 g and major CHD | 22 |
Clinical Features
EA is commonly associated with maternal polyhydramnios, and with increasing frequency, the diagnosis is being made antenatally particularly if there is no TEF. In the postnatal period, symptoms associated with the condition include excessive salivation, feeding difficulties, respiratory distress, and cyanotic episodes. Cases of EA (with the exception of the rare EA with double fistula) can be confirmed by failure of passage of a nasogastric tube into the stomach. Cases of TEF in the absence of EA (i.e., H-type fistula) may present later, usually with recurrent episodes of respiratory distress or pneumonia.
Treatment
The overall aim of surgical correction is early division of any fistula with the respiratory tract to protect the lungs and airway, and restoration and maintenance of esophageal continuity to allow normal feeding. Following diagnosis, a Replogle tube is placed in the upper esophageal pouch which allows suction of secretions and minimizes the risk of pulmonary aspiration. Surgical repair involves ligation and division of any fistula and primary anastomosis of the two ends of the esophagus where possible. Infants who are too unstable to tolerate primary esophageal repair, typically preterm infants with respiratory distress, may be treated with ligation of the fistula only followed by delayed esophageal anastomosis once a period of cardiorespiratory stability can be achieved. Most infants with “pure EA,” that is, EA without TEF, and some infants with EA with TEF have a gap between the two ends of the esophagus that is too wide for primary anastomosis to be achieved. This group poses a particular surgical challenge. These infants are typically fed by gastrostomy initially, and the esophageal anastomosis is re-attempted after a period of growth (typically at least 6 weeks) during which the esophageal ends often grow closer together permitting anastomosis. If attempts at anastomosis remain unsuccessful, esophageal replacement such as by gastric transposition is considered.
Outcome
The majority of patients do well following anastomosis, but a number of complications may occur, and they require recurrent procedures. The most common surgical complication is anastomotic stricture. Strictures frequently require dilatation, and balloon dilatation is the preferred technique. Other complications include anastomotic leakage, recurrent TEF, gastroesophageal reflux, and disordered peristalsis. The combination of poor esophageal motility and gastroesophageal reflux with or without anastomotic stricture may lead to long-term feeding difficulties.
The Stomach
The most common abnormality of the stomach in the neonatal period is hypertrophic pyloric stenosis . Whether this is truly a congenital abnormality or an acquired disorder is questionable. It is further discussed in Chap. 4 .
Other congenital conditions affecting the stomach including congenital microgastria, gastric volvulus, and congenital gastric outlet obstruction due to a pyloric web or atresia are all extremely rare and are mentioned only for completeness.
Obstructive Lesions of the Duodenum, Jejunum, and Ileum
The most common congenital conditions affecting the duodenum, jejunum, and ileum all result in partial or complete gastrointestinal obstruction. The presenting features and investigations recommended to diagnose the underlying abnormality are similar for all conditions. The clinical features and investigations of these conditions are therefore presented first followed by a description of each type of abnormality and the recommended treatment options.
Clinical Features
Obstructive lesions of the small intestine from the pylorus down to the ileocecal valve may give rise to polyhydramnios in the antenatal period which is detectable by antenatal ultrasonography. As a general rule, the more proximal the lesion the more severe the degree of polyhydramnios, and distal ileal lesions may be present in the absence of polyhydramnios [5]. The list of differential diagnoses giving rise to polyhydramnios is however extensive. Another feature that may be seen on antenatal ultrasonography is the presence if dilated loops of intestine with or without echogenic bowel. While the combination of echogenic and dilated loops of bowel is often a sign of some form of intestinal abnormality, the precise nature of any problem is rarely identified before birth. One exception to this is a diagnosis of congenital duodenal obstruction in which case the appearance of a double bubble on antenatal ultrasonography has high sensitivity and specificity.
Following birth, the most common and important clinical manifestation of obstructive lesions of the GIT is bile-stained vomiting. Vomiting with truly bilious staining is always abnormal in the neonatal period and always requires investigation. Lesions in the duodenum and jejunum usually result in bilious vomiting within hours. In addition, the abdomen may appear empty or even scaphoid, and visible gastric peristalsis may be observed. Lesions lower in the ileum result in a distended abdomen if the obstruction is complete, and there may be failure to pass meconium. Obstructive lesions may also give rise to intestinal perforation in the neonatal period and occasionally antenatally. In all cases of neonatal intestinal obstruction , infants become progressively hypovolemic and are prone to circulatory and respiratory collapse. They require fluid resuscitation and may require ventilatory support. GIT obstruction should therefore be considered in any infant who is dehydrated especially if there is a history of vomiting.
Stenotic lesions of the small bowel in which the obstruction is incomplete may give rise to increased diagnostic difficulty. Affected infants may present with intermittent vomiting and episodes of partial obstruction. They eventually fail to thrive or develop complete obstruction at which stage they are fully investigated and the diagnosis becomes apparent.
Intestinal malrotation is considered separately as it may present with a spectrum of clinical scenarios depending on the degree of intestinal obstruction or midgut volvulus or both. The clinical pictures of all types of abnormal rotation are those of acute or chronic intestinal obstruction and/or acute or chronic abdominal pain suggestive of intestinal ischemia. True malrotation typically presents in the first year of life with symptoms of upper GIT obstruction including vomiting which is usually bile-stained. A coexisting volvulus may be suspected by abdominal pain , peritonitis , and hypovolemic shock associated with intestinal ischemia. However, these signs may be relatively nonspecific in the young infant. Malrotation may also present later in life.
Investigations
The aim of investigating cases of suspected obstruction of the small intestine is twofold. First to identify the nature and anatomical location of the lesion to allow for planning of correct treatment and second to identify cases of malrotation in whom there is a risk of midgut volvulus and intestinal ischemia. These cases require urgent surgical intervention to reduce the risk of potentially catastrophic intestinal necrosis. The history and examination may give clues as to the location of the lesion as described above. An abdominal X-ray may simply confirm the presence of dilated intestinal loops but may also give further clues in some cases. A double bubble appearance on abdominal X-ray with a lack of air in the distal intestine (Fig. 3.2) is characteristic of duodenal obstruction. Multiple air-filled loops of proximal bowel often with air-fluid levels along with a paucity or complete absence of gas in the distal bowel is highly suggestive of obstruction of the ileum. Intestinal perforation if present will usually be apparent on abdominal X-ray, and in the rare cases of antenatal perforation, there may be widespread or localized flecks of calcification representing calcified meconium within the peritoneum.
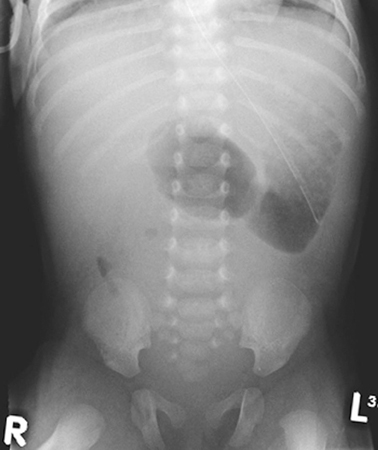
Fig. 3.2
Abdominal X-ray of an infant with duodenal atresia showing the “double-bubble” appearance characteristic of duodenal obstruction. (Reprinted with permission from [4], Fig. 2.2)
In cases in which the diagnosis is not clear on abdominal X-ray or in which midgut malrotation or volvulus is suspected, a limited upper gastrointestinal contrast study is indicated. The classical finding in cases of malrotation is that the duodenojejunal flexure lies to the right side of the spine instead of its normal left-sided position (Fig. 3.3). This finding should prompt urgent surgical treatment due to the risk of coexisting midgut volvulus. The contrast study may also identify the presence of a stenotic segment or complete obstruction.
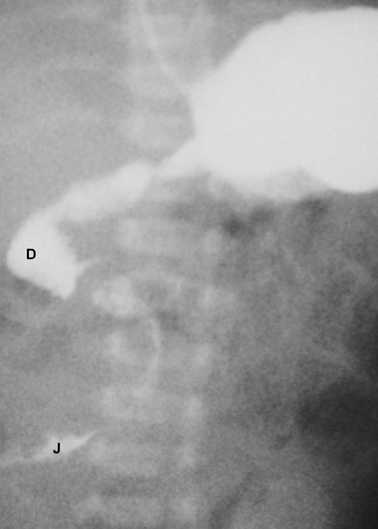
Fig. 3.3
Upper gastrointestinal contrast study of a case of malrotation. The contrast is seen within the duodenum ( D) and flowing into the upper jejunum ( J) both of which lie completely to the right of the midline. (Reprinted with permission from [4], Fig. 2.3)
Cases of lower ileal stenosis or atresia are often more difficult to diagnose, and a contrast enema is invaluable in distinguishing between ileal and colonic obstruction and could be therapeutic in cases of meconium ileus (see below) .
Conditions Affecting the Duodenum
Duodenal atresia and duodenal stenosis both of which may be associated with an annular pancreas are the commonest congenital conditions to affect the duodenum. Both are capable of giving rise to duodenal obstruction. The incidence is reported to be between 1 in 5000 and 10,000 live births [6].
Explanations of the etiology of duodenal atresias are not universally accepted. Unlike atresias of the ileum, they are not thought to be due to vascular accidents, and the most widely accepted explanation is that of failure of recanalization of the intestinal lumen during early embryonic development.
Classification
There are four basic types of duodenal obstruction (Fig. 3.4). In type 1, there is a stenosis of the duodenum resulting from a diaphragm or web partially or totally occluding the lumen. Due to the incomplete nature of the obstruction, cases may present in childhood rather than in the neonatal period. In type 2 duodenal atresia, the proximal and distal segments end blindly but remain connected by a fibrous cord. There is complete separation of the bowel segments in type 3 and type 4 comprises an atretic segment with an annular pancreas. Multiple atresias are said to occur in up to 15 % of cases [7].
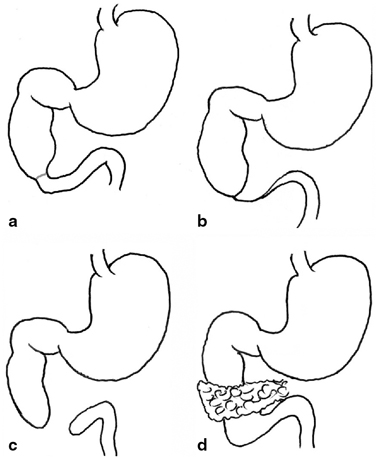
Fig. 3.4
Variants of duodenal atresia. a type 1 atresia due to an internal diaphragm, b type 2 atresia with blind-ending loops remaining connected by a fibrous cord, c type 3 atresia with blind ends completely separated, d type 4 duodenal obstruction with an annular pancreas. (Reprinted with permission from [4], Fig. 2.4)
Treatment
The principles of treatment are to restore intestinal continuity while avoiding interference with the ductal system draining the pancreas and biliary tree. This is best achieved using a duodenostomy in which the obstructed segment is bypassed by joining the proximal segment directly to the distal segment. Following surgery, the long-term gastrointestinal results are good [8].
Conditions Affecting the Ileum and Jejunum
The main congenital problems directly affecting the small intestine from the duodenojejunal flexure down to the cecum are atresia and stenosis . Jejunoileal atresia occurs more commonly than its duodenal counterpart with an incidence of varying from 1 in 330 to 3000 live births [9]. Such lesions are one of the most common causes of neonatal intestinal obstruction . The major difference between atresias of the ileojejunum and those of the duodenum is in their etiology. It is postulated that atresia or stenosis of the jejunum and ileum is the result of a localized vascular accident during intrauterine life. Subsequent ischemic necrosis and reabsorption of the affected segment or segments result in a contracted scarred bowel wall leading to stenosis at one end of the spectrum to a complete intestinal and mesenteric defect at the other. Fetal animal experiments have confirmed at least in part this hypothesis [10], and the absence of other congenital abnormalities found in association with jejunoileal stenoses and atresias supports the localized vascular accident theory.
Classification
Morphological classification of these lesions allows different surgeons and centers to compare outcomes, and it is also of therapeutic and prognostic value. The most commonly accepted system is that proposed by Louw [11] and modified by Grosfeld [12]. Whether the lesion is classified as ileal or jejunal is determined by the most proximal affected segment (Fig. 3.5).
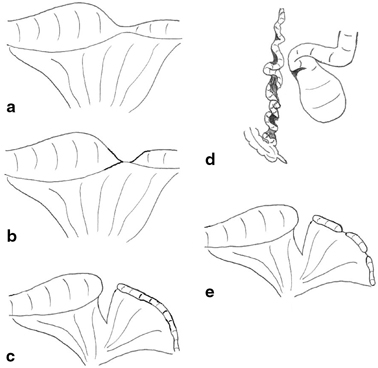
Fig. 3.5
Variants of ileal atresia . a type I due to an internal web (not shown) with no mesenteric defect, b type II atresia with blind ends joined by a fibrous cord, c type III(a)—blind ends separated with a mesenteric defect, d type III(b) in which the ileum is coiled like an “apple peel” around a single vessel and completely separated from the proximal dilated jejunum, e type IV or multiple atresias. (Reprinted with permission from [4], Fig. 2.5)
Stenosis is a localized narrowing of the lumen without any break in the continuity or mesenteric defect. The intestinal wall may be thickened and rigid at the stenotic site, and there is a small, often minute lumen. The overall intestinal length is not shortened. Type I atresia is the result of a membranous web occluding the lumen with no mesenteric defect and no intestinal shortening. The lumen is usually completely occluded, and the proximal bowel therefore dilated but remaining in continuity with the collapsed distal segment. Type II atresia arises from a complete obliteration of the intestinal segment into a fibrous cord which joins two blind ends and runs in the free edge of the mesentery. There is no mesenteric defect, and once again, the total bowel length is usually normal. In both type III(a) and III(b) atresia, the intestine is likely to be shortened, and this may have significant clinical consequences. Type III(a) atresia consists of blind-ending proximal and distal bowel with no connection and an often large mesenteric defect. The blind ends are often physiologically abnormal with decreased or absent peristaltic activity which may give rise to torsion, distension, or perforation. Type III(b) atresia, also known as apple-peel atresia because of its gross morphology, may involve massive intestinal loss. It consists of intestinal atresia near the ligament of Treitz, obliteration of the superior mesenteric artery (SMA) beyond the origin of the middle colic branch, and the absence of the dorsal mesentery. The remaining intestine is coiled helically (like an apple peel) around a single perfusing vessel, often has impaired vascularity, and is almost inevitably short. Furthermore, there may be additional segments of type I or II atresia within the apple-peel segment. Such a configuration most likely arises from occlusion of the SMA due to thrombus, embolus, or strangulation as part of a midgut volvulus. In type IV atresia, there are multiple atretic segments, and the intestine may resemble a string of sausages. Overall, bowel length is usually shortened, and the intestine grossly dilated. It has been proposed that the etiology of type IV atresia may be due to failure of recanalization of solid epithelialization throughout the length of the intestine rather than from multiple single vascular events.
While it is generally accepted that stenosis and atresia of types I, II, and III(b) are the result of intrauterine vascular accidents, a genetic component has been suggested in type III(b) and IV.
Treatment
The mainstay of surgical treatment for this type of lesion is resection of the atretic or stenotic segment and primary anastomosis with closure of the mesenteric defect. The proximal intestinal segment is usually dilated and functionally abnormal with absent or ineffective peristalsis. This dilated proximal segment is excised along with a short segment distal to the stenosis or atresia. It is essential to establish patency of the distal bowel by irrigation or wash out intestine, and subsequently, a primary anastomosis is performed. There is a balance to be struck between the length of dilated proximal segment resected and the risk of leaving the infant with a short length of small bowel. As such, it is almost inevitable that the caliber of proximal bowel will be greater than that of the distal intestine, and a number of techniques exist to assist construction of the anastomosis in such circumstances.
Outcome following intestinal atresia is dependent primarily on the length of remaining intestine and the presence of the ileocecal valve. Short bowel syndrome has been defined as the presence of less than 75 cm of the small intestine or 30 % of the predicted intestinal length in a premature infant [13, 14]. Outcomes following short bowel syndrome vary, and there is a high level of dependence on parenteral nutrition. However, intestinal adaptation can occur such that more than 80 % of babies with short bowel syndrome do eventually become entirely enterally nourished [15].
Intestinal Malrotation
The incidence of intestinal malrotation is difficult to truly establish as not all affected patients develop symptoms, but autopsy studies estimate the incidence at approximately 1 in 500.
The traditional embryological basis for disorders of intestinal rotation is that of abnormal positioning of the intestinal loops in relation to one another as they return to the abdominal cavity from the yolk sac. During normal development, the midgut rotates through 270° so that the duodenum lies posterior to the colon, and the duodenojejunal flexure is to the left of the midline. A consistent finding in cases of malrotation is abnormal positioning of the duodenojejunal flexure. However, an alternative hypothesis has been proposed based on animal studies [16]. Kluth proposes that malrotation is the result of failure of localized growth of the duodenal loop rather than a disorder of rotation.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


