Etiology of liver cirrhosis
Metabolic liver disease
α1-Antitrypsin deficiency
Wilson disease
Progressive familial intrahepatic cholestasis
Glycogen storage diseases
(especially types IV, VI, IX, and X)
Lipid abnormalities (e.g., Gaucher disease)
Peroxisomal disorders (e.g., Zellweger syndrome)
Tyrosinemia
Cystic fibrosis
Developmental/ genetic:
Biliary atresia
Congenital hepatic fibrosis
Alagille syndrome
Infections:
Viral (e.g., chronic hepatitis B or C)
Parasitic (e.g., echinococcosis)
Immune related:
Autoimmune hepatitis
Sclerosing cholangitis
Vascular:
Budd–Chiari syndrome
Hepatic veno-occlusive disease
Portal vein thrombosis
Miscellaneous:
Nonalcoholic steatohepatitis (NASH)
Drugs
Pathogenesis of Liver Fibrosis
Persistent insult to either hepatocytes or cholangiocytes acts as a stimulus for fibrosis to set in. Hepatotropic viruses, toxins, and diseases that cause chronic cholestasis (tyrosinemia, progressive familial intrahepatic cholestasis, etc.), damage liver cells, either by apoptosis or necrosis leading on to activation of a cascade of events causing fibrosis/cirrhosis (Fig. 71.1). The damaged liver cells secrete inflammatory mediators such as transforming growth factor (TGF)-beta1, TNF-α, epidermal growth factor (EGF), insulin-like growth factor (IGF), endothelin (ET), and platelet-derived growth factor (PDGF) that activate HSC. These damaged hepatocytes/cholangiocytes also recruit and activate T cells and Kupffer cells which secrete interleukin 6 (IL-6), interferon (IFN)-alpha, CD40, CCL21, and IGF which in turn activates HSC via a different pathway. The ECM such as type IV collagen, fibrinogen, and urokinase-type plasminogen activator that is secreted during the process of fibrosis could act as a stimulus for further activation of HSC starting a vicious cycle [3].
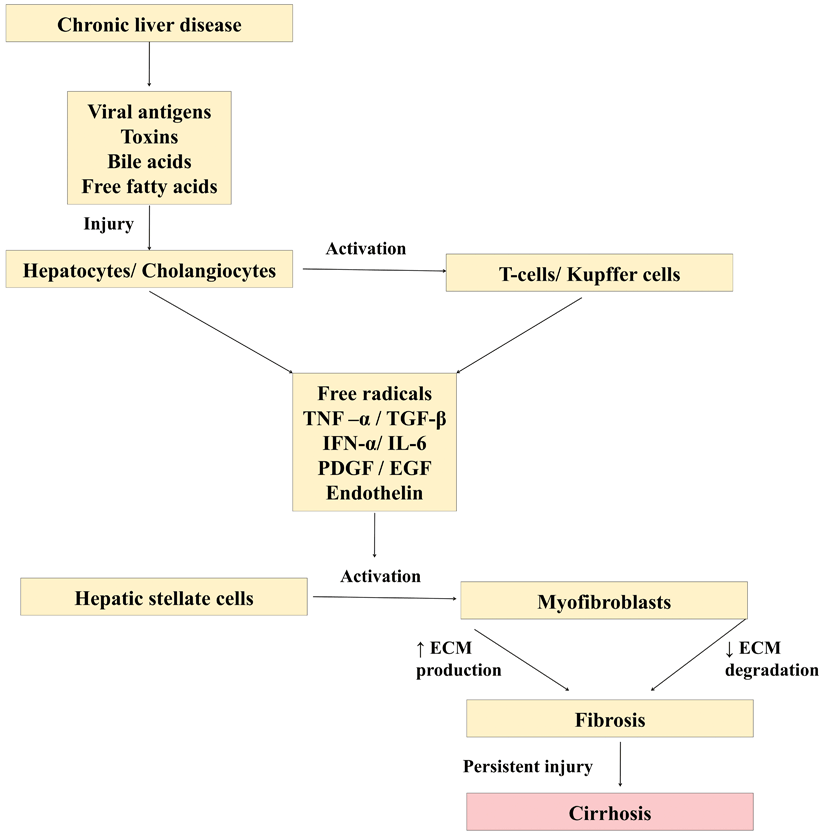
Fig. 71.1
Diagram showing cascade of events leading on to liver cirrhosis
Under normal condition, HSC are quiescent cells that store lipids and vitamin A in liver that resides in space of Disse. When activated, HSC transdifferentiate to myofibroblast-like cells, acquiring fibrogenic and secretory properties. Apart from HSC, myofibroblasts that are derived from small portal blood vessels and bone marrow play a role in ECM formation during fibrosis of liver [4–6]. The source of fibroblast that is involved in fibrosis of liver depends on the etiology of insult to the liver [6]. With biliary pathology such as biliary atresia, portal tract fibroblasts, and in hypoxic insults, bone marrow fibroblasts play an important role [7, 8].
The ECM laid during this process of fibrosis differs in structure and composition, when compared to the ECM laid during routine remodeling. Mitogenic stimuli such as TGF-beta upregulate several precollagenous genes and increase the secretion of collagen by myofibroblasts, particularly type 1 and 3. Activated HSC also enhance the secretion of ECM-digesting enzymes like interstitial collagenase, gelatinase A, and stromelysin-1 which degrade specific components of ECM such as type 4 collagen and laminin, paving way for increased composition of type 1 and 3 collagen in fibrosis [9]. Increased tissue inhibitors of matrix metalloproteinases (TIMPs), which inhibit collagenase, result in gross imbalance between production and degradation of ECM. The fibrous tissue thus formed could interconnect portal tracts, portal tracts and central venules, or a mixture of both patterns and progress to cirrhosis. Contrary to the initial belief, it is shown that liver fibrosis even cirrhosis could regress to some extent on successful treatment of underlying cause [10].
Diagnosis of Liver Fibrosis
Fibrosis of liver covers an entire spectrum of microscopically detectable fibrosis during initial stages to macroscopically gross fibrosis as in advanced stages of CLD. There is no validated biochemical test or scoring system based on biochemical tests that would grade the severity of fibrosis [11]. Synthetic liver function or cholestasis has no correlation with the degree of fibrosis, as these parameters may be normal even in advanced fibrosis as in congenital hepatic fibrosis. Imaging techniques such as ultrasonography, computer tomography, and magnetic resonance imaging (MRI) can identify liver fibrosis though quantification is difficult. Liver biopsy is considered to be the gold standard method for the assessment of liver fibrosis as it provides necroinflammatory and the fibrosis grade. Figure 71.2a shows complete nodule formation, and Fig. 71.2b shows extensive collagen, when stained by trichrome in a cirrhotic liver. Scoring of degree of fibrosis could be done using Metavir (stages I–IV) and Ishak score (stages I–V). Due to invasive nature of the procedure, it is practically difficult to monitor fibrosis progression using liver biopsy. To overcome this problem, transient elastography (FibroScan®), a noninvasive technique is used in serial monitoring of progress of fibrosis with good accuracy and has been validated even in children [12] .
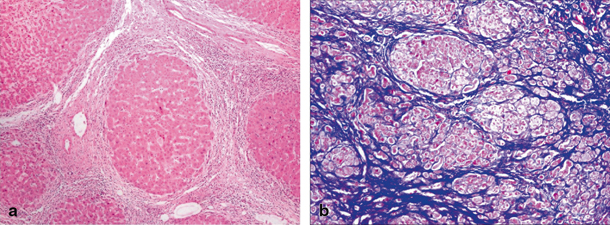
Fig. 71.2
a Microscopic image of cirrhotic liver showing a nodular formation. Hepatic lobule ( arrow head) completely encased by fibrous tissue. b Microscopic image of trichrome-stained liver, where collagen is stained blue, while the cytoplasm of hepatocytes is stained red and nuclei as black structures within cells
Complications and Management of Cirrhosis
CLD and the term “cirrhosis” have been used interchangeably in literature as cirrhosis is a manifestation of CLD, and most of the complications in CLD are secondary to cirrhosis. With progressive fibrosis, there is distortion of blood vessels and bile leading on to increased resistance to blood flow resulting in increased portal pressure on afferent side and tissue hypoxia due to reduced blood supply on efferent side. Cirrhosis also leads to increase in hepatic artery resistance, and in the case of hypotension secondary to an infection or variceal bleed, the liver could suffer hypoxic insult and could decompensate. Over a period of time, the fibrous tissue coalesces to form a tight capsule and restricts hepatocyte regeneration and worsen cholestasis. Synthetic/detoxification failure, cholestasis, and fibrosis individually or in combination contribute towards complication of CLD, some of which are highlighted in Table 71.2 . There is an increased risk of hepatocellular carcinoma (HCC) in cirrhotic liver, and the generalized nodular transformation of liver makes it difficult to diagnose HCC at early stages. Ultimately, these patients would succumb either to liver failure or to its complication unless treated appropriately. Fibrosis is a reversible phenomenon, but the extent to which the distorted liver architecture would revert back to its original state still remains an unanswered question. Disease-specific treatment, for example, chelation for Wilson’s disease and steroids for autoimmune liver diseases, halts the progress of fibrosis and could reverse cirrhosis. At present, there is no approved drug that would inhibit or reverse fibrosis.
Table 71.2
Signs and symptoms of cirrhosis and probable mechanism causing it
Complications of cirrhosis | ||
|---|---|---|
Symptoms | Signs | Underlying mechanisma |
Jaundice, pruritus | Elevated bilirubin and bile acids | Detoxification failure of liver |
Altered mental status | Encephalopathy | Detoxification failure of liver |
Distended abdominal veins, vomiting blood | Caput medusae | Portal hypertension |
Abdominal distension Swelling of legs | Ascites/edema | Hypoalbuminemia, portal hypertension |
Muscle wasting | Malnutrition | Decreased absorption of nutrients due to defective bile flow and increased demand |
Telangiectasia | Spider nevi | Detoxification failure of certain hormones |
Easy fractures | Osteoporosis and osteomalacia | Hepatic osteodystrophy |
Decreased urine output | Oliguria | Hepatorenal syndrome |
Bluish discoloration and swelling of fingers | Cyanosis and clubbing | Hepatopulmonary syndrome, portopulmonary hypertension |
Difficulty in breathing | Dyspnea | Hepatopulmonary syndrome, portopulmonary hypertension |
Complications of Cirrhosis
Portal Hypertension
Liver is a unique organ with dual blood supply , via the hepatic artery and portal vein . The portal vein, formed by the splenic vein and superior mesenteric vein, supplies nutrient-rich blood from the intestine to the liver. The usual pressure is less than 5 mmHg, and this blood traverses hepatic sinusoids reaching hepatic venules having much lower pressures and enters systemic venous circulation. PHT develops when there is increased resistance anywhere along the portal system, and thus PHT could be extrahepatic (portal vein thrombosis), intrahepatic (cirrhosis), or posthepatic (hepatic venous thrombosis). PHT leads on to opening up of porto-systemic collaterals causing varices, which could rupture and cause bleed .
Direct measurement of portal pressure is difficult, and so hepatic venous pressure gradient (HVPG) is measured via hepatic venous catheterization. HVPG is pressure difference between wedged hepatic venous pressure (WHVP) and free hepatic venous pressure, which is reflective of portal pressure. HVPG of more than 10 mmHg is associated with the development of varices, and pressures more than 12 mmHg is associated with variceal bleed. In clinical practice, accurate measurement of portal pressure by invasive methods is of little relevance as esophageal varices and splenomegaly hint towards the development of PHT, and this needs to be managed appropriately .
Pathogenesis of PHT in Cirrhosis
Like any other tubular organ, resistance and flow govern the pressure in portal venous system. In CLD , there is a substantial increase in both flow and resistance of portal vein leading on to net increase in portal pressure.
Increased Resistance to Portal Flow
In cirrhosis , dynamic contractile elements within sinusoids and portal venules contribute up to 40 % of increased vascular resistance, while the rest is attributed to fibrosis. Wrapping of HSC and myofibroblasts can wrap around sinusoids, and the portal venular smooth muscle contraction can increase vascular resistance [13]. ETs are a group of peptides, which cause vasoconstriction and stimulation of cell proliferation in tissue. ET-1 and ET-3 are found to be increased in cirrhotics when compared to controls, probably due to increased production [14]. Angiotensin II, norepinephrine, and thromboxane A2 (TXA2) are other vasopressors which are found to be increased in cirrhosis of the liver and cause increased vascular resistance of portal venules [15]. There is an associated endothelial dysfunction, which further increases the vascular resistance instead of decreasing it. This is probably due to increased levels of TXA2 impairing the response to endothelium-dependent vasodilator acetylcholine, and thus the resultant increased vascular resistance is probably due to imbalance between increased vasoconstrictors and decreased vasodilators in CLD [16].
Increased Portal Circulation
Splanchnic vasodilatation combined with hyperdynamic circulation contributes to increased portal circulation. Nitric oxide (NO) [17] increases the production of cyclic guanosine monophosphate, which directly relaxes the smooth muscle. Increased NO due to enhanced endothelial NO synthase (eNOS) activity in splanchnic circulation is suggested to play an important role in splanchnic vasodilatation. Increased levels of glucagon found in cirrhosis cause direct vascular smooth muscle relaxation and decrease the sensitivity of vascular smooth muscle to endogenous vasoconstrictors. Other vasodilators such as capsaicin–calcitonin gene-related peptide, neuropeptides, and adenosine have a doubtful role. Plasma volume expansion due to sodium retention in cirrhosis also plays a role in increased portal flow.
Variceal Bleed in Portal Hypertension
Apart from ascites and hypersplenism, the major complication of PHT is variceal bleed. HVPG of more than 10 mmHg results in opening up of porto-systemic collaterals , which are situated at the lower end of esophagus , rectum, paraumblical, and retroperitoneal regions. Life-threatening bleeds are usually from esophageal and gastric varices that arise from a collateral network through the coronary and short gastric veins draining into the systemic azygous vein. Miga et al. showed that 20 % of children had esophageal variceal hemorrhage (EVH) within 2 years after Kasai portoenterostomy, and the risk of death or need for liver transplantation was 50 % at 6 years after the initial episode of EVH in a cohort of children with biliary atresia [18]. Etiology plays an important role in the survival outcome after variceal bleed, with varices due to portal vein thrombosis has better prognosis when compared with those due to intrinsic hepatobiliary disease [19].
Management of Variceal Bleed
Variceal bleed is a life-threatening emergency, and the patient needs to be stabilized before shifting for emergency endoscopy. In the event of hypovolemic shock, fluid resuscitation has to be initiated while awaiting whole blood, and in addition, fresh frozen plasma/platelets/cryoprecipitate/factor VIIa might be required, as these patients would be coagulopathic secondary to liver disease. Nasogastric tube placement and gastric lavage help to quantify the blood loss as well as to remove blood from the stomach that could precipitate encephalopathy. Vasoactive drugs that decrease portal pressure (Table 71.3) should be started along with proton pump inhibitors/H2 blockers and antibiotics. Once stable, endoscopic variceal ligation (EVL) or endoscopic sclerotherapy could be done [20]. In the case of continuous bleed and hemodynamic instability, balloon tamponade with a Minnesota or Sengstaken–Blakemore tube should be attempted. Both these tubes have esophageal and gastric balloon, but Minnesota tube has esophageal aspiration port in addition to gastric aspiration port while Sengstaken–Blakemore tube has only gastric aspiration port. These tubes need to be placed by an experienced person as improper placement or overinflation of the balloon can result in esophageal rupture and/or ischemia [21]. Once bleeding is controlled, endoscopic sclerotherapy (ESL) and endoscopic variceal ligation (EVL) has to be performed, as conservative management (medication and balloon tamponade) without endoscopic intervention was associated with 3.6 times higher risk of rebleed [22].
Table 71.3
Various drugs used in portal hypertension and its mechanism of action
Medications used in portal hypertension | ||
|---|---|---|
Drugs used in acute variceal bleed | ||
Drug | Dosage | Mechanism of action |
Vasopressin | Initial IV bolus 0.3 U/kg (maximum: 20 U) over 20 min followed by infusion: 0.002–0.01 U/kg/min | Acts via V1 receptors, reduces portal blood flow, portal systemic collateral blood flow, and variceal pressure |
Octreotide | 1 μg/kg/h IV Infusion or 2–4 μg/kg/dose/8 h s.c. | Analogue of somatostatin Reduce portal flow and pressures |
Terlipressin | 8–20 μg/kg of body weight given at intervals of 4–8 h | Terlipressin, a prodrug of vasopressin |
Drugs used to prevent rebleed | ||
Propranolol Atenolol | 1–5 mg/kg/day in three divided doses 1 mg/kg/day in two doses (Dose has to be adjusted to decrease heart rate by 25 %) | Reduces portal hypertension by decreasing cardiac output and inducing splanchnic vasoconstriction by blocking β-1 and β-2 receptors |
Carvedilol | 0.3 mg/kg/day | Nonselective β-blocker with additional α-1 blocking action |
ESL is a procedure where sclerosant such as ethanolamine oleate or sodium tetradecyl sulfate is injected into a varix under direct vision, thereby obliterating the vessel. Cyanoacrylates are synthetic glues used in gastric varices that rapidly solidify on contact with water and blood. Usually, cyanoacrylates are mixed with ethanolamine oleate at 1:1 ratio to decrease the rate of solidification, as it minimizes the inadvertent adherence to catheters and endoscope. EVL is a procedure where an elastic O-ring is applied over a small area of esophageal mucosa and submucosa, causing strangulation of the tissue (varix) caught in-between the ring, eventually leading to fibrosis and obliteration. Randomized trial of endoscopic sclerotherapy versus variceal band ligation for esophageal varices showed that while both are equally effective in controlling acute bleed; EVL achieved variceal obliteration in fewer treatment sessions along with significantly lower rate of the development of portal gastropathy and rebleeding [23]. Meta-analysis suggests that pharmacotherapy coupled with either EVL or ESL showed better initial bleeding control and 5-day hemostasis, but no effect on mortality [24]. Occasionally, bleeding from gastric varices and Roux en Y jejunal loop varices might be difficult to control and might require interventional measures such as transjugular intrahepatic portosystemic shunt (TIPSS), surgical shunts, esophagogastric devascularization ± splenectomy [25]. Liver transplantation has to be offered to those with CLD, after control of the acute bleeding episode.
Prophylactic Therapy for Variceal Bleed
The American Association for the Study of Liver Diseases (AASLD) practice guidelines recommend β-blockers as first-line therapy in adults with medium/large esophageal varices and EVL for patients in whom β-blockers are contraindicated or poorly tolerated [26]. Trials on children showed that β-blockers and EVL/ESL are relatively safe to be used, but so far, there are no robust data to suggest that its use as primary prophylaxis decreases the incidence of first episode variceal bleed in children [27, 28]. Variceal eradication and nonselective beta-blocker are recommended in adults after an episode of bleed as secondary prophylaxis, while in children, small studies have shown usefulness of EVL/EST to prevent rebleed, but role of beta-blocker is still unclear [29, 30].
Neurological Complications in Cirrhosis
CLD predisposes to a variety of neurological complications such as intracranial hemorrhage due to the presence of coagulopathy, increased risk of cerebral infection due to decreased immune function, and hepatic encephalopathy (HE) [31].
Hepatic Encephalopathy
HE is defined as a metabolically induced, potentially reversible, functional disturbance of the brain that may occur in acute or CLD[32]. Though HE is considered to be a potentially reversible condition, some patients might not recover to their previous level of cognitive function after a severe episode of HE.
Factors such as infection, trauma, and electrolyte imbalance in the background of CLD could cause sudden liver decompensation and lead to HE. Encephalopathy that occurs in acute liver failure (ALF) is categorized as type A, HE in portosystemic shunt without any intrinsic hepatocellular disease as type B , and in CLD as type C. Type C could be further subclassified into minimal, episodic, or persistent. HE is called minimal hepatic encephalopathy (MHE), when diagnosis could be made only on psychometric analysis in an apparently normal person with CLD. Episodic encephalopathy in CLD coincides with episodes of high protein intake, gastrointestinal bleed, infection, etc. Usually, these episodes resolve with the treatment of the precipitating factors, but sometimes they persist and termed as persistent HE.
Clinical Features
HE could be clinically graded from 1 to 4 using West Haven criteria, and Glasgow coma scale which has lesser interobserver variability could be used in assessing conscious levels [33]. MHE is associated with mild intellectual impairment, where verbal ability is preserved and apparently normal personality [34]. Worsening of HE leads on to asterixis (flapping tremor), hypertonia and hyperreflexia, hypotonia, and areflexia may be seen with subsequent progression to coma. Features similar to Parkinsonism such as muscular rigidity, bradykinesia, hypokinesia, monotony of speech, and tremors could be seen in HE.
Pathogenesis of Hepatic Encephalopathy
The pathogenesis of HE is thought to be multifactorial, with neurotoxins and altered neurotransmitters acting on the neurons. Ammonia is considered to be an important factor involved in pathogenesis, while mercaptans, short- and medium-chain fatty acids, phenols, methionine derivatives, etc. play a minor role. altered ratio of excitatory (↓ dopamine and noradrenaline) and inhibitory (↑ gamma-aminobutyric acid (GABA) and serotonin) neurotransmitters resulting in abnormal cerebral function. Apart from these, there is an increased formation of false neurotransmitters such as octopamine, phenyl methionine from accumulation of phenylalanine, and tyrosine. There are other proposed mechanisms of HE such as increase in false neurotransmitters (octopamine, phenyl methionine), altered ratios of branched-chain amino acids (BCAA) and aromatic amino acids (AAA), changes in postsynaptic receptor activity, increased permeability of blood–brain barrier, etc.
Role of Ammonia in HE
Ammonia is produced from a variety of sources such as small intestine enterocytes, gut flora, muscle, and kidney. Under healthy condition, around 80–90 % of ammonia is either converted to urea by periportal hepatocytes or glutamine by perivenous hepatocytes. Though ammonia is implicated in HE, the exact mechanism is still elusive. Few of the suggested mechanisms are: (1) Excess ammonia is converted to glutamine by glial cells. This glutamine increases intracellular osmotic pressure, leading on to swelling of astrocytes, cerebral edema and can competitively bind to glutamate receptors and inhibit them. (2) Oxidative stress triggered by ammonia toxicity in the astrocyte resulting in mitochondrial dysfunction. (3) Enhanced cytokine activity and impaired intracellular signaling [35].
It was thought that ammonia in portal circulation was exclusively produced by the gut flora, but current hypothesis suggests that small gut enterocytes produce ammonia in excess of gut flora, and gut decontamination alone has minimal effect on ammonia levels [36]. Kidney is involved in both production and elimination of ammonia. Kidney can metabolize glutamine to produce ammonia and bicarbonate and can excrete ammonia as ammonium ion and urea in urine [35]. Alkalosis can decrease the conversion of ammonia to ammonium, as there is no need to excrete the hydrogen ion resulting in elevated free ammonia that can cross blood–brain barrier and precipitate HE. Myocytes can act as buffer by converting ammonia to nontoxic glutamine via glutamine synthetase, and thus poor muscle mass is an important risk factor for HE [37].
Role of Neurotoxins and Inflammatory Mediators in HE
The ratio of BCAA/AAA phenylalanine, tyrosine, and tryptophan is called the Fischer ratio [38, 39]. There is decrease in Fischer ratio in liver failure, due to preferential usage of BCAA by muscles and decreased clearance of AAA by liver. Elevated serum AAA can cross the blood–brain barrier into the brain and results in synthesis of false neurotransmitter such as octopamine and synephrine [40].
Several short-chain fatty acids such as propionate, butyrate, and valerate are produced in small intestine by breakdown of proteins by fecal flora [41]. These short-chain fatty acids competitively inhibit urea cycle enzymes and can bind to albumin and displacing albumin-bound toxins, thus precipitating HE. Indole, oxindole, endozepines, neuronal acetylcholinesterase , TNF-α, and allopregnanolone are few of the other bioactive substances that could play a role in HE [42–46].
Diagnosis
Diagnosis of encephalopathy is mainly based on clinical history and examination [47]. Clinical diagnosis of mild-to-moderate HE is difficult in children as it requires their cooperation to perform psychometric tests. Neuroimaging and electrophysiological studies of brain would give supportive evidence rather than to confirm the diagnosis of HE. Though arterial ammonia level correlates with the severity of HE, it could not be used to grade encephalopathy [33]. HE could be clinically graded using West Haven criteria, and conscious level is graded using Glasgow coma scale [33].
Neuropsychological Assessment
In children, neuropsychological assessment should be made using tests such as Wechsler intelligence tests and Dutch child intelligence test (Revisie Amsterdamse Kinder Intelligentie test), where validated nomogram exits for the selected age, sex, racial, and ethnical subgroup. The Psychometric Hepatic Encephalopathy Score (PHES) and number connect test (NCT) A and B could be used to diagnose MHE in adults while these tests are not validated in children [48, 49].
Critical Flicker Frequency
Critical flicker-frequency threshold is a simple and reliable test for the quantification of low-grade HE [50]. The principle of the test is to identify the point of switch over from a steady red light to a flickering light. A normal human eye perceives flicker rate of 60 Hz as a steady light. The frequency of the light is gradually decreased to a point where the patient starts to perceive the light as flicker. Using this technique, it was found that patients with HE can pick up flickering only when the flicker rate comes below 39 Hz, while cirrhotic counterparts without encephalopathy and normal individual could identify flickering at higher rate [47]. The need for understanding (and being able to follow instructions) the test limits is used only in children more than 8 years [51].
Electroencephalogram
Using electroencephalogram (EEG), HE could be graded into 5 grades (0–4). Grade 0 is a normal EEG with regular alpha rhythm. Grade 1 encephalopathy is characterized by irregular background alpha rhythm and appearance of theta rhythm. Theta activity becomes continuous with occasional delta wave in grade 2 HE, and theta activity becomes prevalent with transient polyphasic complexes of spikes and slow waves in grade 3. Grade 4 is deep coma, having continuous delta waves with abundant complexes of spikes and slow waves [47]. With the availability of newer complex EEG analytical softwares, EEG can be analyzed in specific regions of the brain.
Neuroimaging in HE
Computer tomography (CT) of brain is not a useful tool in diagnosis of HE, apart from excluding organic causes such as bleed, tumors, or gross edema that can cause encephalopathy [52]. MRI is a better modality to diagnose cerebral edema and demyelination associated with HE. Conventional MRI lacks sensitivity in diagnosing milder forms of edema, and newer techniques such as magnetization transfer imaging (MTI), fast fluid-attenuated inversion recovery (FLAIR) imaging, and diffusion-weighted imaging are more sensitive in picking up brain tissue water content [53]. Proton MR spectroscopy could measure different levels of metabolites in brain such as myo-inositol, choline, and glutamine These specialized neuroimaging is more of research interest and provides little benefit in routine clinical practice.
Management of Hepatic Encephalopathy
Management should be directed towards stabilization of the patient and treating the precipitating factors such as bleeding, electrolyte imbalance, and infection In HE of grade 3 or 4, elective intubation has to be considered to protect the airways. Despite increased total body water, these patients would be intravascularly fluid depleted. Hydration is best monitored by central venous pressure (CVP) and ideally should be 6–8 cm of H2O. Though their serum sodium is low, they will have high total body sodium, and any attempt to normalize the sodium will lead on to worsening of edema. Supplementation of extra sodium is required if the sodium level falls below 120 mEq/l [54].
Lactulose decreases the colonic pH to around 5, and thereby decreasing the bacterial fermentation and production of short-chain fatty acids [55, 56]. In acidic environment, the ammonia produced in colon is converted to ammonium ion, reducing its diffusibility back into circulation and thereby removed along with stools. The amount of lactulose dose has to be titrated to produce three soft stools/day. Long-term lactulose in CLD helps to prevent HE, but will not change the ultimate requirement of a liver transplantation. Lactitol (B galactosido-sorbitol) is a nonabsorbable sugar which has similar action to lactulose, but less sweeter and more palatable [57].
Neomycin, vancomycin, and rifaximin are few of the antibiotics that are used to eliminate ammoniagenic bacteria from the gut, thereby decreasing the ammonia production in the intestine. Of all the antibiotics, rifaximin has been shown to be very effective in decreasing ammonia production and highest risk benefit ratio [58]. Rifaximin and lactulose given together was found to be more beneficial than either of them given alone [59]. Use of probiotics has shown to reduce plasma ammonia, but there was no change in final outcome [60].
l-ornithine-l-aspartate (LOLA) enhances the action of ornithine and aspartate transaminases in brain and peripheral tissues to produce nontoxic glutamate. There is no standard dosing in children, up to 20 g/day could be given at an infusion rate not exceeding 5 g/h as suggested by drug monogram and not recommended in children less than 8 years old [54]. Sodium benzoate (250 mg/kg/day) along with intravenous sodium phenylacetate (250 mg/kg/day) or oral sodium phenylbutyrate (250 mg/kg/day) could be used to eliminate ammonia by alternate pathways, thereby decreasing serum ammonia levels. Water-soluble hippuric acid and phenylacetyl-glutamine are formed by conjugation of benzoate with glycine and phenyl acetate with glutamine, respectively. Hippuric acid and phenylacetyl-glutamine are then excreted by kidneys, thereby reducing ammonia load [61]. Though all these medications are more useful in hyperammonia due to urea cycle defects, they are used off-label in hyperammoniemia due to liver failure [35].
There has been increasing interest in the possibility of providing an extracorporeal liver support system for short periods while liver allograft becomes available. High cost, nonavailability of small filters, and lack of safety data in children limit the usage of these devises to clinical trial setting. Liver transplantation has shown to be the definitive treatment, which has improved survival outcome both in adults and children. Elective liver transplantation offers 5-year survival of more than 85 % in children. Synthetic liver failure with encephalopathy is one of the indications for liver transplantation in CLD, which has to be done before permanent neurological damage sets in.
Ascites
Fluid retention and low albumin in CLD result in accumulation of fluid in extravascular space leading to edema, ascites , and plural effusion. Presence of ascites is one of the variables that increase the mortality in children with CLD [62]. Onset of ascites indicates decompensation of liver disease and the need for liver transplantation. Ascites increases the risk of bacterial peritonitis and HRS, which potentially adds on to the already increased mortality associated with liver decompensation (see Fig. 71.3).
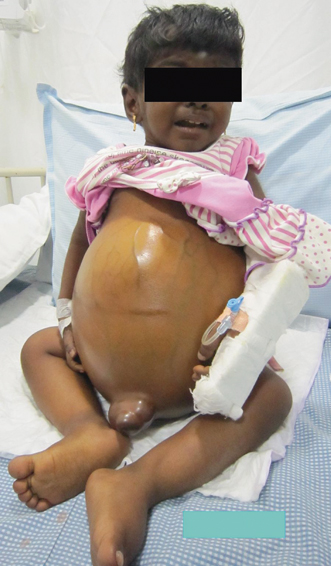
Fig. 71.3
Image of a child with failed Kasai portoenterostomy showing gross ascites, dilated abdominal veins, and edema of legs
Pathophysiology of Ascites
There are several theories based on the triggering factor of the vicious cycle that leads to fluid retention and ascites . The “overfill” theory suggests hepatorenal reflex to be the primary event that leading on to sodium and fluid retention and subsequently ascites. The “underfill” theory suggests that that the increased pressure in portal sinusoids results in increased hydrostatic pressure leading on to excessive lymph production. When lymph production exceeds absorption, the net result is ascites and contraction of intravascular volume (underfill) . “Peripheral arterial vasodilation hypothesis” which was proposed in 1988 suggests that splanchnic and peripheral arterial vasodilatation secondary to PHT and cirrhosis is the initial triggering event [63]. This causes decreased intravascular volume, leading on to baroreceptor-mediated activation of renin angiotensin aldosterone system and release of antidiuretic hormone, resulting in hypervolemic stage due to renal sodium and water retention [63–65]. Splanchnic vasodilatation along with hypervolemia results in increased hydrostatic pressure that ultimately results in passage of fluid to peritoneal space [66]. It is difficult to explain the complex cascade of events leading on to ascites based on one theory, and it is possible that mechanisms as suggested by different theories could contribute to various stages of ascites formation .
Biochemical Diagnosis
Ascitic tap should be carried out in any new onset ascites. Estimating serum-ascites albumin gradient (SAAG) is considered to be superior in making differential diagnosis of probable cause of ascites , compared to quantifying it as exudate or transudate. SAAG ≥ 1.1 g/dL is associated with cirrhosis, portal or hepatic venous occlusion, congestive cardiac failure, etc., while SAAG < 1.1 g/dL is associated with nephrotic syndrome, peritoneal infection, or malignancy [67]. Spontaneous bacterial peritonitis is an important complication of ascites and has to be differentiated from surgical cause of peritonitis such as perforation which accounts for 5 % of cases [68]. Polymorphonuclear neutrophils (PMNs) of > 250 mm3 in ascitic fluid are suggestive of the presence of bacterial infection while predominant lymphocytic cells in ascites (> 20 % of the total leukocyte count) along with ascites–blood glucose quotient of < 0.7 is suggestive of tuberculous infection. In the case of high PMNs in ascitic fluid with symptoms of fever and abdominal tenderness, antibiotics need to be continued even if the cultures are negative as 34.5 % turn culture positive on repeated samples.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


