Fig. 56.1
Schematic illustration of biliary atresia (based on Japanese Association of Pediatric Surgeons classification)
A more radical approach to the technique was pioneered in Sendai, Japan, during the 1950s and 1960s by Morio Kasai (1922–2008) to the problem of “uncorrectability” [2, 3]. He advocated a more radical approach to the biliary dissection and simply transected at the most proximal point in the porta hepatis. The porta, even if there were no visible ducts, was then anastomosed to a Roux loop (portoenterostomy). Transection exposes residual microscopic bile duct remnants within the fibrous tissue which retain communication with the intrahepatic duct system (still often very abnormal). Hence, bile flow actually occurs to a varying degree, but in perhaps the majority, enough to lose their jaundice (see later for results and outcome), postoperative outcome significantly improved.
For the first time, Kasai portoenterostomy (KPE) enabled a much larger cohort of long-term survivors, initially in Japan [3] but later from the 1970s in Europe and North America. It wasnot a cure though, and even survivors displayed many complications related to liver fibrosis and cirrhosis .
Thomas Starzl, in 1963, attempted the first liver transplant in humans in a child born with BA [4]. Sadly, the child died of operative bleeding related to severe portal hypertension. This prompted a number of units around the world to set up transplantation programmes, but, in the absence of effective immunosuppression, they were all terminated by the end of the 1960s. With the discovery of cyclosporine at the beginning of the 1980s, transplantation once more became a viable option, and from this the current strategy of an initial attempt to restore (some might say resurrect) bile flow with a KPE followed by liver transplantation if this fails evolved.
Variants of Biliary Atresia
BA is not a single disease rather it should be thought of as a phenotype resulting from a number of different and entirely separate aetiologies leading to the final common phenotype of biliary inflammation, luminal obliteration and fibrosis [5] .
We are able to define clinically four broad groups (Fig. 56.2):
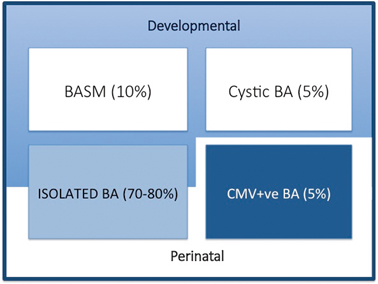
Fig. 56.2
Variants of biliary atresia. BASM biliary atresia splenic malformation, BA biliary atresia, CMV cytomegalovirus, IgM immunoglobulin M
1.
Syndromic biliary atresia. While BA is usually an isolated abnormality found in otherwise normal-term infants, there are a group of infants (about 10–15 % in European and North American series, but < 2 % in Asian series) with other non-biliary anomalies and a poorer prognosis. We have termed this specific constellation of anomalies the biliary atresia splenic malformation (BASM) syndrome [6, 7]. All will have a splenic malformation, usually polysplenia but occasionally asplenia and about half will have situs inversus and congenital heart abnormalities. Other anomalies are evident at laparotomy including preduodenal portal vein, absence of the inferior vena cava and malrotation. Most infants with this syndromic form of BA are female. It is speculated that such cases may result from some fundamental derangement of extrahepatic bile duct (and other systems) development within the embryological phase (< 6 weeks gestation). A genetic aetiology has not been convincingly shown clinically, though several candidate genes exist (e.g. CFC-1). There is some evidence that affected infants have been exposed to an abnormal first-trimester intrauterine environment such as that found in maternal diabetes and thyrotoxicosis.
2.
Cystic biliary atresia (CBA) . In about 5 % of cases, there is obvious cyst (sometimes containing bile) formation within an otherwise obliterated biliary tree. In recent years, it has been possible to detect this cystic change on antenatal ultrasound (US) scans as early as the 18th week of gestation [8, 9]. CBA should not be confused with cystic choledochal malformation (CM) , which can be indistinguishable on US [10]. Discrimination may be made clinically as CBA will invariably have conjugated jaundice, pale stools, etc., and at laparotomy as the cholangiogram will show the abnormal and primitive intrahepatic duct structure. Both CBA and the syndromic BA can clearly be termed “developmental” as whatever caused their pathology occurred during prenatal life.
3.
Isolated biliary atresia (IBA). This is the typical BA variant accounting for about 70–80 % of cases [11]. They have no other significant features and usually display an obliterated biliary tree at laparotomy (usually type 3). In the absence of any clinical or laboratory evidence, we do not actually know the aetiology in this group.
4.
CytomegalovirusIgM +ve biliary atresia. Although there is a range of possible hepatotropic cholangiopathic viruses (e.g. reovirus type 3, rotavirus) it has been difficult to definitively ascribe clinical consequences to infection. We have recently discriminated infants with CMV IgM +ve antibodies from their IgM −ve fellows clinically, histologically and in their response to treatment (unpublished results). This is still controversial and other European series have not shown any such differences; however, the focus in these has been on a group of viruses rather than specifically CMV [12].
Epidemiology
Population-based studies reporting incidence and outcomes of BA are scarce. There is marked variation in incidence across the globe with the highest rates found in East Asian countries compared to Europe and North America [13]. The place with the highest reported incidence appears to be Taiwan at an incidence of 1 in 6622 [14].
The incidence in the UK is about 1 in 17,000–18,000 live births [11], and this appears to be similar to reports from France [15], Sweden [16], Germany [17] and Switzerland [18]. Most large series show an equal gender split although by variant there is marked female predominance in the developmental forms of BA [7, 9, 11, and 15].
Clinical Features
The key features of BA are persistent conjugated jaundice , acholic stools and dark urine in an otherwise healthy term infant (Fig. 56.3). The latter feature is caused by excess conjugated (i.e. water-soluble) bilirubin passing into the urine causing its colour to darken. Such alternative pathways of bilirubin excretion are more developed or at least preserved in the newborn, and very high levels of bilirubin (> 300 μmol/L or > 17 mg/dl) are exceptional. Sometimes jaundice will be difficult to discern in infants of Asian or Afro-Caribbean origin leading to delays in diagnosis and treatment. The median time to referral was 47 days in non-white versus 52 days in white infants in one UK study [21]. Furthermore, all of those “missed” and referred beyond 100 days were non-white.
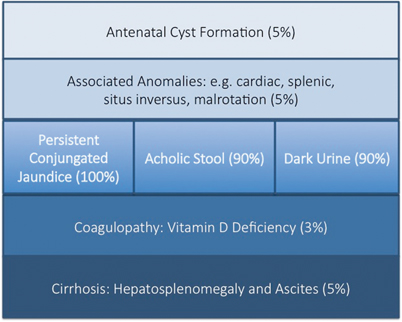
Fig. 56.3
Modes of presentation of biliary atresia
Sometimes the antenatal US scan will be abnormal showing a sub-hepatic cyst, and one should be suspicious of CBA [8]. There is no difference in gestational age or birth weight between those with developmental BA compared to isolated BA, but both groups show a failure to thrive by the time they are admitted. Fat malabsorption is the presumed mechanism and will also cause deficiency of the fat-soluble vitamins D, A, E and K. Low vitamin D levels are common even in those infants presenting early, and this is exacerbated in those of Asian family origin, presumably reflecting low maternal stores (Fig. 56.4). Vitamin K deficiency is possible and a proportion will present with a bleeding tendency, perhaps from the umbilical stump or more catastrophically with an intracranial haemorrhage. Marked elevation of the international normalised ratio (INR) or prothrombin time will be seen. Some syndromic cases will present early because of other abnormalities associated with BASM (e.g. cardiac anomalies, malrotation or situs inversus).
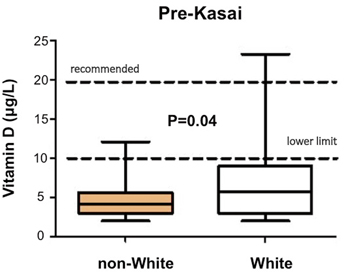
Fig. 56.4
Decreased preoperative vitamin D-levels in non-white compared to white infants with biliary atresia. (Courtesy of Ms. Anu Paul)
Diagnosis
The diagnostic work up in our institution includes a detailed US of the liver, liver biochemistry and a percutaneous liver biopsy [10]. Using this, more than 90 % will have an accurate diagnosis before laparotomy.
Ultrasonography
The US examination is a key part of the diagnostic protocol as it usually excludes other surgical diagnoses (e.g. CM, inspissated bile syndrome, etc.). All of these are characterised by intrahepatic or common bile duct (CBD) dilatation. US may be suggestive of BA as a diagnosis—by showing a shrunken, atrophic gallbladder with no evidence of filling between feeds. In about 20 % of cases, a “normal gallbladder” is described—which turns out to be a mucocele of the gallbadder together with a relatively preserved CBD and an absent CHD; Fig. 56.1).
Laboratory Findings
Liver biochemistry will show a conjugated jaundice (typically > 100 μmol/L), modestly raised transaminases (> 100 μmol/L) as well as significantly raised γ-glutamyl transpeptidase (GGT > 200 IU/L). Serum protein and albumin levels should be normal. However, none of this is specific.
Percutaneous Liver Biopsy
In the authors’ unit, the pre-laparotomy diagnosis of BA is usually made by percutaneous liver biopsy showing histological features characteristic of large duct obstructions such as bile duct proliferation, portal oedema and absence of sinusoidal fibrosis. It is less accurate the younger the infant, and it does require an experienced and confident liver pathologist.
Aspartate Aminotransferase-to-Platelet Ratio Index
The aspartate aminotransferase-to-platelet ratio index (APRi) can be used as a surrogate of liver fibrosis in many liver diseases, including BA. We have recently reported that this correlates significantly with age at surgery and was much higher in CMV-associated BA. Macroscopic cirrhosis evident at laparotomy could also be predicted using a cut-off value of 1.2, with reasonable sensitivity (75 %) and specificity (84 %) in a large cohort of infants from our unit [22].
The usual differential diagnoses of conjungated jaundice include TORCH infections (e.g. toxoplasma, rubella, CMV, hepatitis, etc.), genetic conditions (e.g. α-1-antitrypsin deficiency, Alagille’s syndrome (abnormal “elfin” facies, butterfly vertebrae, pulmonary stenosis), progressive familial intrahepatic cholestasis (PFIC) disorders, metabolic conditions (e.g. cystic fibrosis, galactosemia), and parenteral nutrition and neonatal hepatitis.
Miscellaneous Diagnostic Techniques
Other techniques which have been used include endoscopic retrograde cholangiopancreatography (ERCP), percutaneous cholangiography, duodenal intubation and measurement of intraluminal bile. Unfortunately, magnetic resonance cholangiopancreatography (MRCP) is not detailed enough as yet to confidently diagnose BA and radioisotope hepatobiliary imaging (e.g. using iminodiacetic acid derivates), that shows absence of bile excretion lacks specificity—many being simply “neonatal hepatitis”.
The surgical differential is less common and includes obstructed CM, which usually shows obvious dilated intra- and extrahepatic biliary dilatation; inspissated bile syndrome which usually occurs in the preterm with a precipitating event such as dehydration or haemolysis and spontaneous perforation of the bile duct which usually shows US evidence of bile ascites and a sub-hepatic collection evident on US [10].
Screening
Some countries have adopted a population screening programme for BA. The most well-developed has been that of Taiwan [23], where mothers are issued with colour-coded cards and asked to compare it with their infant’s stool. Recognition of pale stool prompts further investigation and referral. This has certainly shortened their time to surgery—the median age at KPE is now < 50 days and is currently the best achieved anywhere in national terms. The key achievement, we believe, has been the marked reduction in late-presenting infants who have already developed obvious cirrhosis [24].
In a recent study from our institution, skills of health-care professionals in recognising pale stools were assessed. Sadly, the study showed that one third of the abnormal stools were not correctly identified by physicians and nurses who were apparently regularly in contact with jaundiced infants. The authors suggested that distribution of standardised stool colour cards throughout community clinics and outpatient departments might rectify this problem [25].
The simplest solution to early diagnosis remains education, however. It is clear from so many histories that it is often the parents who recognise that persistent jaundice in their infants is not normal but then are falsely reassured by health visitors and their community doctors who fail to inquire about pale stools or dark urine (nevermind looking at them) and fail to do the appropriate blood test (split bilirubin for conjugated and unconjugated fractions). The statutory community check in the UK occurs at 6 weeks of age and is far too late to make a difference in when these affected infants come to surgery.
Kasai Portoenterostomy
The aim of surgery is to excise all extrahepatic biliary remnants allowing a wide portoenterostomy onto a portal plate, denuded of all such tissue (Fig. 56.5). In most cases, this will expose sufficient transected microscopic bile ductules which retain connections with the primitive intrahepatic bile ductule system to allow restoration of at least a degree of biliary drainage. This should be the object of all three types of BA.
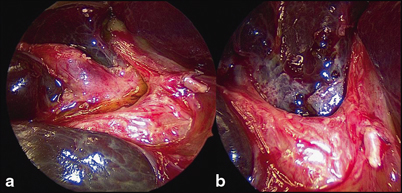
Fig. 56.5
Close-up of porta hepatis during Kasai portoenterostomy. a Hypertrophied proximal biliary remnant (BR) being separated from vascular structures of liver. b Same view following resection of BR, showing denuded portal plate
A short right upper quadrant muscle-cutting incision should be performed initially to confirm the suspected diagnosis or, if the gallbladder is shown to contain bile, to proceed to on-table cholangiography.
There are then two surgical strategies to expose the porta hepatis: One involving division of at least the left-sided suspensory ligaments and then eversion of the liver onto the abdominal wall. The other retains the liver in the abdominal cavity but usually requires dissection and slinging of the right and left portal veins to allow full exposure of the biliary remnants. A simple portoenterostomy using a retrocolic Roux loop (about 40 cm) completes the reconstruction (Fig. 56.6). Older techniques involving stomas in the Roux loop have been abandoned by virtually all centres.
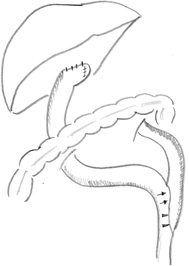
Fig. 56.6
Schematic illustration of retrocolic Roux-en-Y loop, typically measured at 40 cm from portoenterostomy to jejunojejunostomy
The KPE can be replicated laparoscopically. However, most centres which explored and pioneered this technique have subsequently reverted to open KPE [26, 27]. The portal dissection, the key to wide excisional surgery, is not improved by being performed laparoscopically and the reconstruction remains technically challenging. There is obviously a better scar, though the infants remain in hospital for the same length of time and an adhesion-free abdominal cavity for the transplant surgeon, although even the latter has been challenged by a study from Germany [28]. Surgeons in Juntendo, Tokyo, have adopted a different approach to these issues by limiting the scale of the portal dissection, and consciously limiting the transection to a basic oval shape—allowing at least some remnant to remain [29].
Postoperative Management
Intravenous fluids and nasogastric aspiration are continued until return of bowel function (about 3–4 days). Careful monitoring of blood glucose, electrolytes and INR is important in the early phase. Liver biochemistry (including bilirubin) may well worsen in the first week, but, by about the 4th week, there should be a definite fall in bilirubin and consistently pigmented stools in those who will do well. Strict attention to nutritional needs is important and all infants need regular vitamin supplementation. Medium-chain triglyceride formula milk (e.g. Caprilon®; SHS, Liverpool, UK) is advocated to maximise calorie input and facilitate lipid absorption.
Adjuvant Therapy for Biliary Atresia
Although a number of drugs have the potential to improve the outcome of KPE surgery, there has been little published in the way of scientific data to provide credible support for any [30] . Postoperative steroids remain popular: A recent study from the USA, including 516 patients from 42 children’s hospitals reported about a usage rate of 46 % [31]; however, there is only a single prospective, double-blind, randomised, placebo-controlled trial using a low dose (2 mg/kg/day) [32]. This showed a significant reduction in early bilirubin levels (especially in young livers) in the steroid group but did not translate to a reduced need for transplant or improved overall survival. A further follow-up study using a higher starting dose of prednisolone (5 mg/kg/day) confirmed biochemical benefit and also reported a 15 % improvement in jaundice clearance when infants < 70 days were selected out [33]. There does not seem to be significant side effects from the high-dose regimens.
Ursodeoxycholic acid (UDCA) also remains popular and may be beneficial, but only if surgery has already restored bile flow to a real degree. Willot et al. from Lille in France assessed the effect of UDCA on liver function in children > 1 year post-KPE and showed that it improved biochemical liver function in stable children [34]. It may also have an extra-beneficial effect in BA because of its immunosuppressive properties as it has been shown to decrease proliferation of and cytokine production by mononuclear cells in vitro [35] .
Complications
About 20–30 % of infants will have no effect from KPE, their stools will remain pale; their bilirubin levels will continue to rise. These infants will inexorably continue to develop cirrhosis and end-stage liver disease with severe failure to thrive, ascites , splenomegaly, deepening jaundice and often bleeding from variceal formation. Such infants need expedited liver transplantation, often before their first birthday. It is important to recognise these infants early and not offer false hope that they will somehow turn a corner—they will not.
Other specific complications deserve a more detailed coverage.
Cholangitis
Re-establishment of bile drainage exposes the child to the risk of ascending cholangitis, which occurs most commonly in the year following primary surgery in about 40–50 % of children. Paradoxically, it only occurs in children with some degree of bile flow, not in those with early failure as described above. The usual organisms are enteric in origin (e.g. Escherichia coli, Pseudomonas, Klebsiella spp.).
Clinically, an episode is characterised by fever, acholic stools and a change in biochemical liver function (rising bilirubin and aspartate aminotransferase; AST levels). The diagnosis may uncommonly be confirmed by blood culture or rarely by percutaneous liver biopsy, but, the key component is antibiotic treatment on suspicion and not confirmation. Intravenous broad-spectrum antibiotics effective against Gram-negative organisms (e.g. piperacillin and tazobactam, gentamicin). Most will respond within 24 h and liver function is usually restored fairly quickly. Some children sustain repeated cholangitis, and they should be treated by prolonged courses of intravenous antibiotics via an indwelling vascular device. If, however, it is clear that there are other features of end-stage liver disease, then they too should be considered for transplant.
Occasionally, cholangitis occurs as a late event in otherwise normal children or adolescents , who have good liver function and have cleared their jaundice [36]. The Roux loop may be at fault here with partial obstruction leading to bile stasis. A combination of radioisotope scans and percutaneous cholangiography may aid the diagnostic process and operative Roux loop revision may be required. Recently, we have used an enteroscope to investigate these patients and provide radiological and endoscopic visualisation of the proximal Roux loop [37].
Portal Hypertension
Increased portal venous pressure has been shown in about 70 % of all infants at the time of Kasai operation [38] . However, subsequent portal hypertension depends on both the degree of established fibrosis and, most importantly, the response to surgery. There is a relationship with biochemical liver function and variceal development and in those who fail and need early transplantation; about 30 % will have had a significant variceal bleed.
Infants and children with bleeding oesophageal varices need rapid access to high-quality pediatric facilities with the resources and expertise to manage them appropriately. Injection and/or banding is not a technique for the occasional pediatric endoscopist. Restoration of circulating blood volume and pharmacotherapy (e.g. 2 ml/h of 500 μg octreotide in 40 ml of saline) should precede endoscopy and achieve a measure of stabilisation. Sometimes a modified Sengstaken tube needs to be placed to achieve control of bleeding. Invariably, in children, this can only be done under general anaesthesia but can be life saving. The definitive treatment in older children is endoscopic variceal banding, although injection sclerotherapy retains a role in treating the varices in infants.
In common with other large centres, we therefore recommend that for each child with BA there is the opportunity to enter a programme of endoscopic surveillance to try and pre-empt variceal bleeding [39]. In this respect, there may be a role for selection based on haematological, biochemical or US variables to assign risk. Our recent work has suggested that APRi and clinical prediction rule (CPR) [40] appear to be superior in this respect to simple univariate indices (e.g. platelets, bilirubin) or US dimensions (e.g. spleen size or resistance index).
The key variceal signs that should prompt prophylactic endoscopic treatment are the presence of significant red wales in grade II/III oesophageal varices and obvious (usually lesser curve) gastric varices [39]. Liver transplantation needs to be actively considered as definitive treatment for portal hypertension where liver function is poor and the child is already significantly jaundiced.
Ascites
This is related to portal hypertension in part, but there are other contributory factors including hypoalbuminaemia and hyponatraemia. It also predisposes to spontaneous, bacterial peritonitis. Conventional treatment includes a low-salt diet, fluid restriction and the use of diuretics particularly spironolactone . It is often seen in settings of malnutrition and end-stage liver disease, and a nutritional supplementation is important to try and increase calorie and protein intake.
Outcome Following Kasai Portoenterostomy
There are many factors, which will influence surgical outcome in BA. Some are unalterable (e.g. degree of cirrhosis at presentation, absence of, or paucity of bile ductules at the level of section) and some are subject to change (e.g. efficacy of the KPE due to surgical inexperience, poor choice of technique, complications postoperatively due to inexperienced unit, untreated cholangitis, etc.). In large centres with experienced surgeons, about 50 % of all infants should clear their jaundice and achieve a normal (< 20 μmol/L or < 1.5 mg/dL) bilirubin [41]. These should do well and have a good quality of long-term survival with their native liver.
There is no doubt that increasing age at KPE is associated with increasing liver fibrosis and cirrhosis although the actual rates of progression differ according to underlying cause [42]. There is a marked relationship with age at surgery for instance with cystic BA and BASM but it is not that evident in isolated BA. So, it is not really possible to use age as a simplistic predictor in individual cases as even in those coming to surgery at > 100 days may still have a response to KPE [43]. Still there may be a role for those with the signs of evident cirrhosis (e.g. ascites, heterogeneity of the liver appearance on US) for consideration of transplantation as the primary procedure. It remains an uncommon choice though, perhaps < 5 % in our experience [41].
In the England and Wales, we have adopted a policy of centralisation of surgeons and resources. So for a country of 56 million, there are only three recognised centres to treat this condition. All surgical facilities including transplantation are available. This policy was adopted because previous audits of outcome had shown a significant difference depending on centre experience with the less experienced centres showing poor outcomes [44]. Subsequent studies confirmed an improvement in overall national outcome [41, 45]. This policy has been replicated by some European countries again with demonstrable benefit [46, 47].
Choledochal Malformation
Introduction
The German anatomist Abraham Vater recognised the ampullary nature of the junction of biliary and pancreatic ducts which since then has carried his name. Subsequently, he also described what appeared to be a choledochal cyst in a pamphlet published in 1723 [48].
Further examples of this, the classical choledochal cyst, were described, but still barely 90 cases had been recorded by 1959, when Alonso-Lej et al. attempted a simple classification into three types [49]. Even as recently as 1975, Flanagan could only identify details of 955 cases from the literature [50].
CM (of which some can be described as choledochal “cysts”) may be characterised as, . “an abnormal dilatation of the biliary tract, in the absence of any acute obstruction”. This allows us to exclude the dilated CBD secondary to choledocholithiasis and strictures secondary to chronic pancreatitis for instance. Similarly, while many CMs present with jaundice and biliary obstruction, it is obvious that previously their function was unimpaired for much of the subject’s life in the presence of clear morphological change.
Aetiology
Most CMs appear in some way to be of congenital origin though the actual mechanism itself is obscure.
There are two competing hypotheses:
1.
Pancreatic reflux. An intrinsic part of most examples of CM complex is an abnormal pancreatobiliary junction. Normally, the pancreatic and bile ducts open separately within the wall of the duodenum at the ampulla of Vater achieving biological separation of bile and pancreatic juice. In most patients with CM, duct confluence occurs within the head of the pancreas, outside the duodenal wall resulting in a common channel that allows free intraductal mixing of both types of secretion [51–53].
Donald Babbitt, an American radiologist, proposed that this reflux of presumably activated pancreatic juice could damage the wall of bile duct causing weakness and dilatation [54]. There are a number of experimental animal models which have tried to replicate the effects of pancreatic enzymes on bile ducts [55, 56], but there has been little actual documented change in the dimensions of the biliary system.
2.




Distal bile duct stenosis. Almost 15 % of CM can now be detected antenatally on the maternal US scan. Most of the infants are not actually jaundiced at birth—though some are. In all of these, the morphological type is a cystic malformation and in these, though there might be a common channel, there is minimal amylase in bile (implying no reflux) and often a very definite abrupt change and distal bile duct stenotic segment. Furthermore, animal models involving ligation of the distal bile duct [57] produce obvious cystic change.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
