Chapter 17 Wilson disease and related disorders
1 The Wilson disease (WD) gene is located on chromosome 13 and encodes a copper-transporting P-type adenosine triphosphatase (ATPase) protein, which is expressed primarily in the trans-Golgi network of the hepatocyte.
2 An impaired or deficient WD gene product is responsible for the lack of copper incorporation into ceruloplasmin and the defective biliary excretion of copper in WD.
3 Most patients with symptomatic WD present with hepatic or neuropsychiatric features; the principal hepatic manifestations include fulminant hepatic failure, chronic hepatitis, and cirrhosis.
4 In patients with a low serum ceruloplasmin level, the diagnosis of WD in the absence of Kayser–Fleischer rings requires determination of the hepatic copper concentration.
5 The use of DNA marker studies is limited largely to genetic screening of young family members or difficult diagnostic situations, with use of the index patient’s DNA as a reference.
6 The drug of choice for treating WD remains D-penicillamine, but alternative drugs under selected circumstances include trientine, zinc, and tetrathiomolybdate. The use of combination therapy (e.g., trientine and zinc) appears promising.
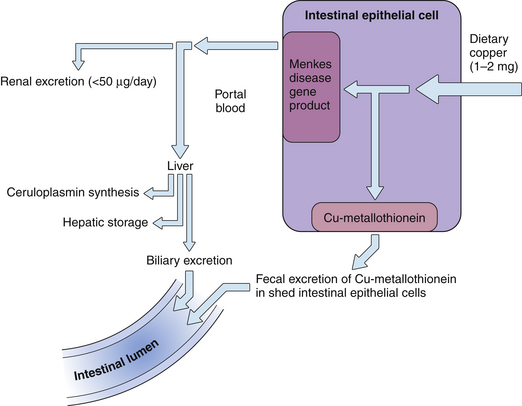
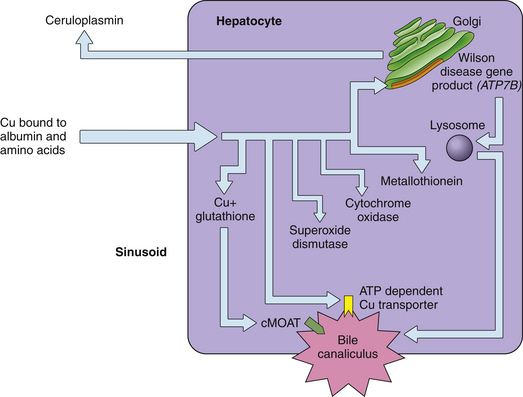
Copper Metabolism (Figs 17.1 and 17.2)
1. Dietary copper (1 to 2 mg/day) is actively transported into the proximal small intestinal epithelial cells.
2. A fraction (25% to 60%) of copper is absorbed and transferred into the portal circulation, bound to serum albumin and amino acids. The remaining intraepithelial copper is bound to metallothioneins and is subsequently excreted as intestinal epithelial cells are sloughed. No significant enterohepatic circulation of copper occurs.
4. Only a small fraction of serum albumin–bound (nonceruloplasmin) copper is normally excreted by the kidney (less than 50 μg/24 hours); most is taken up by hepatocytes.
5. In the hepatocyte, copper is complexed with and detoxified by metallothioneins or glutathione and is used as a cofactor for specific cellular enzymes, incorporated into ceruloplasmin, or excreted into bile.
6. The site of copper incorporation into ceruloplasmin may be the Golgi apparatus. The WD gene (ATP7B) product, also known as the Wilson adenosine triphosphatase (ATPase), is presumed to be responsible for copper transport in this compartment and subsequent incorporation into ceruloplasmin.
7. The delivery of copper to specific intracellular locations is mediated by small cytosolic proteins termed copper chaperones.
8. As the copper content of the hepatocyte increases, the Wilson ATPase transfers from the trans-Golgi network to a vesicular compartment adjacent to the canalicular membrane.
9. Biliary excretion of copper occurs partly through a vesicular pathway. Another, perhaps less important, route of excretion is as copper-glutathione. A third potential excretory pathway is through a specific copper transporter in the plasma membrane.
Genetics
1. WD is an autosomal recessive disease with a gene frequency of 0.3% to 0.7%, thus accounting for a heterozygote carrier rate of slightly greater than 1 in 100. In 1985, the WD gene was shown to be linked to the red cell enzyme, esterase D, an association that established the location on chromosome 13.
2. In 1993, three different groups of investigators isolated the WD gene by using positional cloning. The gene, designated ATP7B, spans an 80-kb region of the chromosome and encodes a 7.5-kb transcript that is expressed primarily in the liver, kidney, and placenta.
3. The gene product is a 1466-amino acid protein, a member of the cation-transporting P-type ATPase subfamily, and is highly homologous to the Menkes gene (ATP7A) product and the copper-transporting ATPase (cop A) found in copper-resistant strains of Enterococcus hirae.
4. More than 269 disease-causing mutations of the WD gene have been identified to date. Most of the mutations are missense mutations. Comparatively few patients are homozygotes for the same mutation; however, most are compound heterozygotes (i.e., bearers of two different alleles).
 Despite the clinical diversity of the disease, allelic heterogeneity at the ATPB7 locus does not appear to account for the marked clinical variability observed in patients.
Despite the clinical diversity of the disease, allelic heterogeneity at the ATPB7 locus does not appear to account for the marked clinical variability observed in patients.
Despite the clinical diversity of the disease, allelic heterogeneity at the ATPB7 locus does not appear to account for the marked clinical variability observed in patients.
Pathogenesis
1. Copper toxicity plays a primary role in the pathogenesis of this disorder. Affected organs invariably exhibit elevated copper levels.
2. Maintenance of normal copper homeostasis depends on the balance between gastrointestinal absorption and biliary excretion. Intestinal copper absorption in patients with WD does not differ from that of physiologically normal or cirrhotic subjects.
3. Biliary excretion of copper is reduced in WD. Studies indicate a possible defect in the entry of copper into lysosomes, but with normal delivery of lysosomal copper to bile. Investigators have suggested that copper transport into the trans-Golgi apparatus by the WD gene product is essential for its routing and excretion through the lysosomal pathway. This process, in turn, depends on the normal recycling of the WD protein between the trans-Golgi network and the vesicular compartment.
4. Deficiency of the plasma copper protein ceruloplasmin is unlikely to have a role in the pathogenesis of WD. The low serum ceruloplasmin level in patients with WD is believed to be the result of a lack of incorporation of copper into apoceruloplasmin, which has a shorter half-life than copper-bound ceruloplasmin.
5. Excess copper appears to exhibit toxicity by the generation of free radicals, which result in lipid peroxidation, depletion of antioxidants, and polymerization of Cu-thionein leading to necrosis and apoptosis. Morphologic abnormalities from oxidant damage have been identified, particularly in mitochondria (i.e., enlargement, dilatation of cristae, and crystalline deposits).
Clinical Features
Hepatic
1. Cirrhosis
 This often occurs early in the course; symptoms may be minimal or absent, with nearly normal liver biochemical tests.
This often occurs early in the course; symptoms may be minimal or absent, with nearly normal liver biochemical tests.
This often occurs early in the course; symptoms may be minimal or absent, with nearly normal liver biochemical tests.

2. Chronic hepatitis
 Young patients may present with features that are indistinguishable from viral or autoimmune chronic hepatitis.
Young patients may present with features that are indistinguishable from viral or autoimmune chronic hepatitis.
Young patients may present with features that are indistinguishable from viral or autoimmune chronic hepatitis.







Neurologic
3. Common early symptoms are dysarthria, clumsiness, tremor, drooling, gait disturbance, masklike facies, and deterioration of handwriting.
4. Rigidity with overt parkinsonian features, flexion contractures, grand mal seizures, and spasticity are seen less often and in the later stages of the disease.
6. Neurologic symptoms may improve markedly with treatment, although residual deficits are common despite adequate chelation therapy.



Psychiatric
1. One third of all patients with WD may present with psychiatric symptoms; patients may be mistakenly diagnosed with a progressive psychiatric illness.
2. Psychiatric symptoms are present in virtually all neurologically affected patients, and the severity tends to parallel that of the neurologic abnormalities.
3. Early symptoms in teenagers may be limited to subtle behavioral changes and deterioration of academic and work performance.
4. Patients present later with personality changes, lability of mood, emotionalism, impulsive and antisocial behavior, depression, and increased sexual preoccupation.










Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


