, Mark Thomas1 and David Milford2
(1)
Department of Renal Medicine, Birmingham Heartlands Hospital, Birmingham, UK
(2)
Birmingham Children’s Hospital, Birmingham, UK
Abstract
In this chapter we explain:
How to draw a family pedigree
Genetic disorders of the glomerulus and tubules
Current understanding of cystic kidney diseases
How vesico-ureteric reflux can be associated with kidney disease
It is well known that some kidney diseases are inherited. It is less well known that someone with a family history of kidney failure has double the normal adult risk of kidney disease from a different cause. Their risk of developing chronic kidney disease between 10 and 19 years of age is six times greater [1]. So it is important to ask about any family history of kidney disease and to document the family in a pedigree chart.
Our understanding of the environmental and genetic factors that lead to this increased risk is advancing rapidly. With modern molecular techniques, genetic testing is relatively simple. However, interpretation of the results and the implications they have for someone’s physical and psychological health can be more complex.
Knowing that you have inherited a kidney condition may allow you to prepare psychologically and physically for kidney failure. Alternatively it may cause prolonged anxiety about an uncertain prognosis. It may affect your ability to obtain insurance or a mortgage. Informing other family members that they may be at risk and advising whether or not they should be genetically tested is a delicate matter.
If someone has a disease caused by a mutation in a single gene, the predictive value of the mutation, the penetrance, is nearly 100 %. However, the disease may appear at different ages and with different severity. Furthermore, some single-gene diseases vary due to interaction between the abnormal disease gene and a mutation in a modifier gene.
Diseases caused by multiple mutations on different genes are much more common. They have a variable phenotype and are more affected by environmental factors. Genome-wide association studies have identified genes that confer an increased risk for hypertension and diabetic nephropathy, as well as vesico-ureteric reflux , focal segmental glomerulosclerosis (FSGS) and atypical haemolytic uraemic syndrome (aHUS) (see Sect. “Kidney disease linked to causes of anaemia” in page 176) [2].
Genetic abnormalities are like experiments of nature. They have helped us to understand how the kidneys work at a molecular level. We will describe the more common disorders in sequence along the nephron from the glomerulus to the ureter.
The Glomerular Filtration Barrier
Alport Syndrome
Cecil Alport described a syndrome of hereditary nephritis with haematuria and deafness in a British family in 1927 [3]. He thought the nephritis was due to an infection but we now know that it and the deafness are due to a mutation of one of the type IV collagen genes. The abnormal collagen disrupts the structure of membranes in the glomerulus (see Fig. 7.1) and in the organ of Corti in the cochlea.
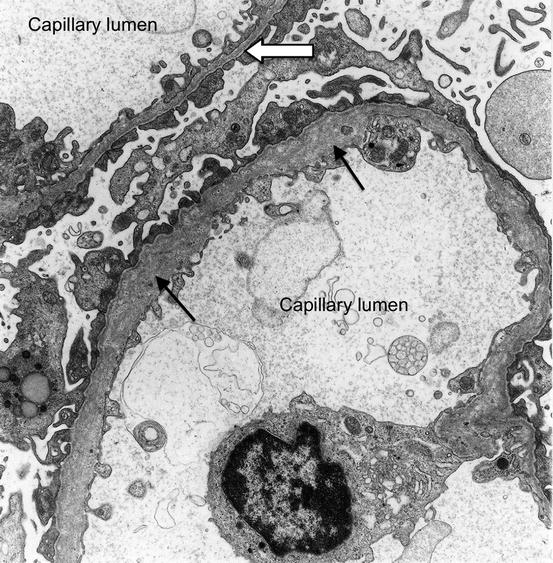
Fig. 7.1
Electron micrograph of a section through two glomerular capillaries in a child with Alport syndrome. The glomerular basement membrane in one capillary is abnormally thin (white arrow). The other is abnormally thick and the arrangement of collagen fibres is disorganised (black arrows) showing a ‘basket-weave’ pattern
Alport reported that: “The male members of a family tend to develop nephritis and deafness and do not as a rule survive. The females have deafness and haematuria and live to old age.” The type IV alpha 5 collagen gene is located on the X-chromosome, explaining why males are severely affected whereas female heterozygotes are less so.
The presentation of nephritis in males can vary. As Alport reported: “The last couple of cases are interesting evidence of the variations in the course of nephritis. The kidneys of two members of the same family were attacked, apparently by the same organism, at the same time, and under exactly the same conditions. The elder brother contracted acute nephritis, which cleared up completely; the younger developed parenchymatous nephritis, passing into small white kidney, and ending in death.”
Alport did not refer to the eye abnormalities that can accompany the syndrome: a misshapen lens (anterior lenticonus ) and abnormal colouration of the retina. These rarely lead to loss of vision [4].
A number of conditions have been described within the Alport syndrome family, involving mutations of genes coding for different protein chains that together form type IV collagen. They are mostly X-linked recessive. Some have autosomal recessive inheritance – homozygotes have the syndrome whereas heterozygote carriers are unaffected or have thin glomerular basement membrane nephropathy (see Patient 11.1). A small number show autosomal dominant inheritance, heterozygotes being affected.
Congential Nephrotic Syndrome
Numerous genetic abnormalities, both dominant and recessive, have been described in children with congenital nephrotic syndrome. The age of onset depends upon the gene affected and whether one or both alleles are affected. If the presentation is before 3 months of age, the cause is invariably genetic [5]. The nephrotic syndrome is steroid-resistant and often progresses to renal failure (see Patient 9.1).
The gene mutations lead to abnormal proteins in the basement membrane or the podocytes and have helped to identify the role of these proteins in the glomerular filtration barrier . For example, in the Finnish type of congenital nephrotic syndrome there is an abnormality of nephrin , a transmembrane protein in the slit diaphragm between the podocyte foot processes. Other causes are mutations in the genes coding for laminin and podocin (Fig. 7.2).
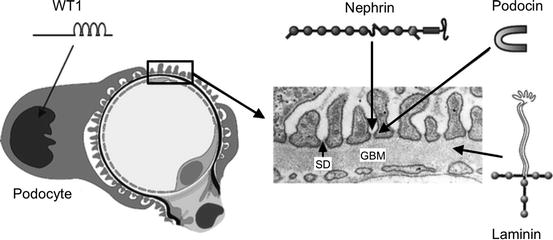
Fig. 7.2
Cross-section of a glomerular capillary (left) and electron microscopy image of a normal capillary wall (right). WT1 is a transcription factor important for podocyte function. Nephrin is a major component of the slit diaphragm (SD) connecting podocyte foot processes. Podocin is an adapter protein located intracellularly in the SD area. Laminin is a major structural protein of the glomerular basement membrane (GBM). Genetic mutations in these proteins lead to congenital nephrotic syndrome (From Jalanko [5])
The Tubules
Figure 7.3 summarises the main genetic diseases affecting the nephron.
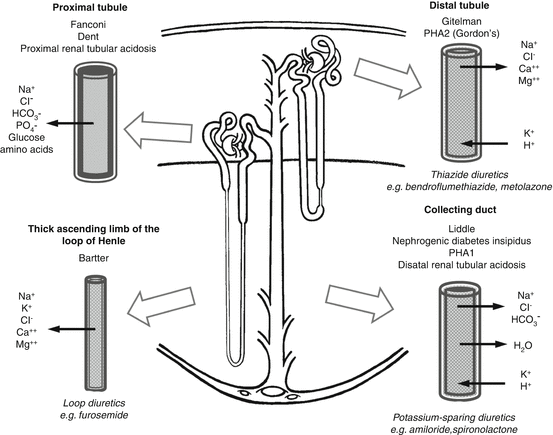
Fig. 7.3
Genetic diseases of the nephron, the normal transport functions that are affected and the sites of action of the main classes of diuretics [6]
The Proximal Tubule
The majority of the reabsorption and secretion of solutes occurs in the proximal tubule [7]. The surface area of the tubule is maximised by the brush border microvilli on the epithelial cell surface. The main features of the genetic disorders that disrupt solute transport are polyuria and the loss of electrolytes, sugar or amino acids into the urine. Table 7.1 describes some of the more common disorders.
Table 7.1
Genetic disorders of the proximal tubule, their mode of inheritance and clinical consequences
Solute | Gene symbol | Gene product(s) | Inheritance | Disorder | Consequence |
|---|---|---|---|---|---|
Glucose | SLC5A1 SLC5A2 | Na/Glu cotransporter SGLT1 SGLT2 | AR | Renal glycosuria | Positive urine dipstick test for glucose |
Amino acids e.g. cystine | CSNU1 SLC7A9 | amino acid transporter | AR | Cystinuria | Cysteine stones |
Bicarbonate | CA2 SLCA4A | Carbonic anhydrase NaHCO3 cotransporter | AR | Proximal (type 2) | Metabolic acidosis |
Multiple solutes | CLCN5 | Cl− channel | XR | Dent type 1 disease : Fanconi syndrome (uricosuria, glycosuria, phosphaturia, aminoaciduria, low molecular weight proteinuria) and proximal renal tubular acidosis | Nephrocalcinosis , stones, osteomalacia , rickets |
Phosphate | PHEX SLC34A3 | Endopeptidase which regulates FGF23 NaP-cotransporter | XD AR | Hypophosphataemic rickets | Phosphaturia, vitamin D resistant rickets |
The Loop of Henle
The loop of Henle is the part of the nephron which enables urine to be concentrated and water conserved [8]. In the thick ascending limb, sodium, potassium and chloride ions are reabsorbed by transporter proteins but water cannot follow [9]. Ions are trapped within the interstitium of the medulla, creating a concentration gradient. The hyperosmolar environment in the deep medulla draws water out of the collecting duct.
Mutations of the genes that code for transporter proteins lead to a condition called Bartter syndrome . The effects are similar to the action of loop diuretics such as furosemide , which blocks the transport of sodium, potassium and chloride into the cells of the thick ascending limb.
Abnormally large amounts of sodium ions flow from the loop of Henle into the distal tubule where they are reabsorbed in exchange for potassium and hydrogen ions. Hence the patient, usually a child, has polyuria, hypovolaemia, hypokalaemia, hypochloraemia and metabolic alkalosis.
Because calcium and magnesium normally are reabsorbed with sodium and chloride in the thick ascending limb, there is also hypercalciuria and hypomagnesaemia.
There are five types of Bartter syndrome with varying renal and extrarenal manifestations [10].
The Distal Tubule
The main function of the distal convoluted tubule is the reabsorption of sodium and chloride, as well as some calcium and magnesium [11]. The sodium/chloride transporter is blocked by thiazide diuretics such as bendroflumethiazide. An autosomal recessive mutation that leads to loss of its function causes similar effects, a condition called Gitelman syndrome [10]. Compared to Bartter syndrome, the salt loss and volume contraction is less severe. The condition usually presents in older children and adults when hypokalaemia and metabolic alkalosis are detected incidentally on a U&E result. In contrast to Bartter syndrome and in line with the effect of thiazides, calcium excretion is reduced.
The opposite of Gitelman syndrome is Gordon’s syndrome (pseudohypoaldosteronism type 2, PHA2 ), an autosomal dominant condition in which the sodium/chloride transporter is overactivated. Excessive sodium reabsorption results in high blood pressure. Reduced potassium and hydrogen ion excretion leads to hyperkalaemia and metabolic acidosis. Thiazide diuretics effectively counteract the abnormalities in Gordon’s syndrome.
The Collecting Duct
The collecting duct has two cell types:
The principal cells contain membrane channels that selectively allow sodium and water to enter the cell and potassium to leave. Sodium is then pumped out through the basal membrane by the Na/K ATPase. An autosomal dominant mutation prevents the epithelial sodium channel being removed from the cell membrane leading to increased absorption of sodium and excretion of potassium and H+. This results in high blood pressure, hypokalaemia and metabolic alkalosis – Liddle syndrome – which is counteracted by amiloride , a potassium-sparing diuretic .
The sodium and potassium channels and the Na/K ATPase in the basal membrane are regulated by aldosterone. Its overall effect is to increase sodium reabsorption and potassium excretion. Mutations of the gene coding for the sodium channel make it unable to respond to aldosterone – pseudohypoaldosteronism type 1 (PHA1) . This results in the opposite of Liddle syndrome – salt wasting, hyperkalaemia and acidosis, like Addison’s disease.
The water channel (aquaporin 2 ) is controlled by the antidiuretic hormone , vasopressin , acting on its receptor. Mutations in the genes that code for the vasopressin receptor or for the water channel lead to hereditary nephrogenic diabetes insipidus . Because the ability to concentrate the urine is lost, the patients have polyuria and nocturia. Urine osmolality is low and plasma osmolality inappropriately raised. This stimulates thirst, leading to polydipsia and preventing severe hypernatraemia.
The intercalated cells break down carbonic acid to H+ and bicarbonate via a reaction catalyzed by carbonic anhydrase. The H+ ions are actively pumped into the urine by an H+ ATPase. The bicarbonate ions are carried to the blood by a Cl−/HCO3 − anion exchanger. Genetic mutations causing loss of function of either of these mechanisms leads to distal (type 1 RTA). The H+ ATPase is also present in the inner ear, hence autosomal recessive mutations cause distal RTA with congenital sensorineural deafness.
The inappropriately alkaline urine in distal RTA leads to the precipitation of calcium in the urine as stones or within the kidney tissue as nephrocalcinosis . In adults, this syndrome is more commonly an acquired defect associated with autoimmune diseases.
Uromodulin
Formerly known as Tamm-Horsfall protein , uromodulin is the most abundant protein in normal urine. Its function is not well understood [14]. It is secreted by the thick ascending limb of the loop of Henle and the distal tubule, where it modulates the sodium and potassium channels. It may also protect against urinary tract infections, ischaemia and stone formation.
Mutations in the uromodulin (UMOD) gene lead to Uromodulin Kidney Disease, an autosomal-dominant condition only fully characterised in the last decade. Genome-wide association studies have also identified the UMOD gene as a risk factor for CKD and hypertension.
Mutations in the UMOD gene alter the structure of uromodulin, preventing its release from the kidney cells and allowing leakage back into the interstitial space around the tubules. In this location uromodulin has an inflammatory effect, leading to interstitial nephritis and fibrosis. End-stage kidney failure is reached between 30 and 60 years of age (Patient 7.1).
Affected kidneys are also unable to excrete uric acid normally. This leads to hyperuricaemia and the early onset of gout .
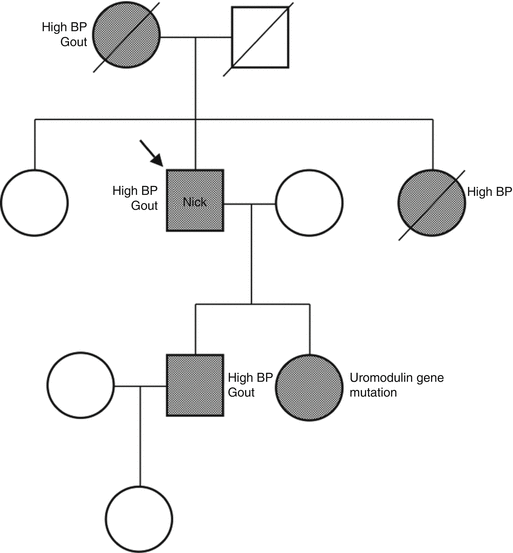
Patient 7.1: Uromodulin Kidney Disease
Nick had a kidney transplant 15 years ago. At that time the cause of his kidney failure was unclear. He had severe hypertension throughout his 20’s and had not been at all keen on having a kidney biopsy. His late mother had gout from the age of 16 followed by severe hypertension, renal failure and a transplant. A renal biopsy had shown normal glomeruli, hypertensive changes in the vessels and some atrophy of the collecting tubules.
Nick’s sister also had severe hypertension and renal failure and had sadly died from complications after a kidney transplant. Neither Nick nor his sister had gout.
On this occasion, Nick attended clinic with his son, aged 19, who was having occasional attacks of gout. His blood pressure was 159/107 mmHg and his eGFR was 53 ml/min/1.73 m2. His urine contained no blood or protein. His serum urate was 769 micromol/L (normal range 200–430).
The association of teenage gout, hypertension and progressive CKD pointed to uromodulin kidney disease. The family had samples taken for genetic testing, which confirmed that Nick and his son and daughter had all inherited a mutation of the uromodulin gene.
Nick’s family pedigree chart is shown in Fig. 7.4.