Fig. 12.1
Uric acid metabolism. Purines are degraded to hypoxanthine and xanthine. Xanthine oxidase converts hypoxanthine to xanthine and also xanthine to uric acid. Unlike most other animals, humans and great ape cannot metabolize uric acid due to a mutation of uricase
A potential benefit of the uricase mutation in the ancestors of the great apes and humans is likely, as a similar mutation of uricase occurred during the same period in the lesser apes. Different hypotheses have been proposed for what the potential benefit might have been to have a higher serum uric acid level. Earlier hypotheses were that uric acid might function as a circulating antioxidant, or potentially as a neurostimulant, that might increase reaction time or performance. More recently, there has been evidence that uric acid may have a role in maintaining blood pressure (BP) and salt sensitivity for our early hominoid ancestors consuming a very low salt diet [3], and also by acting to improve fat stores that might aid survival during periods of famine [4]. Regardless of the mechanism, serum uric acid is higher in humans compared to most other mammals and is also less regulatable and more sensitive to change with diet.
Normal serum uric acid levels vary between 3 and 7 mg/dL in humans. Uric acid levels begin to increase during adolescence years in males but not until menopause in females due to increased uric acid excretion in females [5], an effect likely mediated by estrogen compounds. Several factors affect serum uric acid levels in humans. First, diets rich in purines (such as umami-based foods) or fructose (such as table sugar and high-fructose corn syrup) can increase serum uric acid. Uric acid can also be generated endogenously by states associated with increased cell turnover (cancer) or from ischemia (from the breakdown of adenosine triphosphate, ATP). Reduced uric acid excretion may also occur in settings associated with reduced renal function or in conditions such as obesity, hyperinsulinemia, or hypertension (Fig. 12.2).
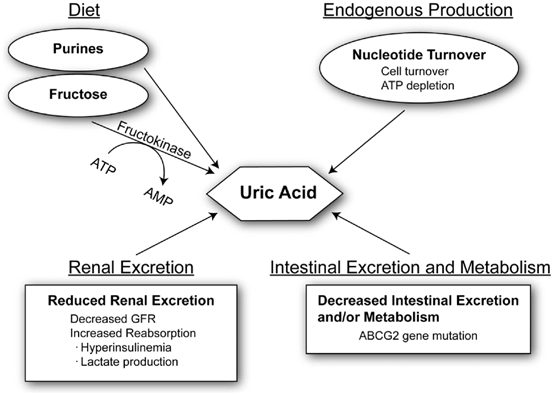
Fig. 12.2
Factors that modulate serum uric acid. Serum uric acid can be modulated by four major mechanisms. Increased production of uric acid can occur from diet or endogenously. Diets rich in purines (umami foods) or fructose can increase serum uric acid. Fructose metabolism by fructokinase leads to degradation of ATP to AMP. AMP accumulation stimulates AMP deaminase, resulting in uric acid production. Increased nucleotide turnover generates nucleotide degradation products metabolized to uric acid. Reduced excretion of uric acid can also alter blood levels. Reduced urinary excretion may occur from reduced glomerular filtration or enhanced tubular absorption. Decreased intestinal excretion and/or metabolism by gut microbiome may alter uric acid levels. ABCG2 stimulates intestinal excretion of uric acid and if mutated can raise uric acid levels. ATP adenosine triphosphate, AMP adenosine monophosphate, GFR glomerular filtration rate, ABCG2 ATP-binding cassette sub-family G member 2
Uric Acid Is a Causative Factor for Hypertension—Historical Perspective
Earliest evidence linking high BP to uric acid can be traced to the 1800s. The medical resident Mahomed [6], proposed an elevated serum uric acid as one of the important mediators of high BP; this was followed by Haig [7] who also highlighted this association in his publications. Subsequently, Davis [8] published a report discussing uric acid as a toxic substance responsible for high arterial tension in gout. The interest in the field was reignited when a report linking uric acid with cardiovascular disease was published by Gertler et al. in 1951 [9]. The strong relationship between uric acid and hypertension was presented in a study by Cannon et al. in 1966, where hyperuricemia was prevalent in 25–50 % patients with untreated hypertension and in 75 % patients with malignant hypertension or coexistent renal disease [10].
Experimental Studies
Early studies investigating the effects of raising uric acid levels in laboratory animals primarily noted the development of acute kidney injury due to the accumulation of urate crystals in the tubules and interstitium. This was also observed in the uricase knockout mouse. While these early models were helpful from the standpoint of understanding the pathogenesis of tumor lysis syndrome, they were not relevant to studies of human hypertension.
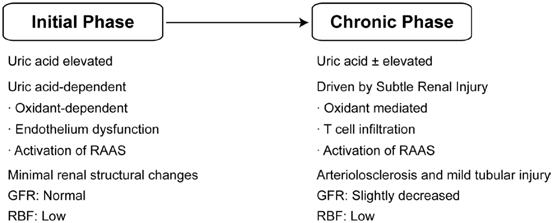
A breakthrough occurred when Mazzali et al. were able to induce mild hyperuricemia by administering an oral uricase inhibitor (oxonic acid) to laboratory rats [11]. The surprising finding was that the rats developed a progressive rise in BP over several weeks that was not associated with any intrarenal crystal deposition or development of renal failure. Indeed, the primary histologic findings were subtle microvascular disease consisting of thickening of the afferent arteriole, not too dissimilar from the renal lesions observed in human essential hypertension. Over time, the increase in uric acid and BP levels was associated with the development of additional histologic changes, including glomerular hypertrophy, tubulointerstitial injury, and low-grade inflammation, in the absence of urate crystals.
An interesting aspect of the animal model was that the hypertension could be divided into two phases. The initial phase was driven by uric acid-mediated activation of the renin angiotensin system, the induction of oxidative stress, and endothelial dysfunction with reduction in endothelial nitric oxide levels. Lowering uric acid levels with xanthine oxidase inhibitor and uricosuric agents in this early phase could completely prevent or treat the hypertension [12, 13]. However, once animals developed significant microvascular injury and tubulointerstitial inflammation, hypertension persisted independent of serum uric acid level in the setting of a high salt diet [3, 14–16].
Clinical Studies
The observation from the experimental studies suggested two phases of hypertension, with the initial phase being uric acid dependent that would occur independent of salt intake, but then converting to a salt-sensitive hypertension driven by subtle injury to the kidney. Interestingly, there is some evidence that this sequence of events may also be observed in human hypertension [14]. Early hypertension (before the age of 40) is often salt resistant (that is, BP is minimally altered by dietary salt intake), whereas hypertension later in life is more commonly associated with salt sensitivity and renal microvascular disease. An elevated serum uric acid has also been repeatedly shown to independently predict the development of hypertension [17]. The strongest association of hyperuricemia is with early hypertension such as that observed in adolescents [18]. Among 125 children (aged 6–18) referred to a tertiary-care renal program for evaluation of newly diagnosed hypertension, 63 had primary hypertension , 40 had secondary hypertension, and 22 had white-coat hypertension. Serum uric acid concentrations > 5.5 mg/dL were found in almost 90 % of subjects with primary hypertension and in 30 % with secondary hypertension. The results were striking, and there was nearly a linear relation between serum uric acid and BP (Fig. 12.3).
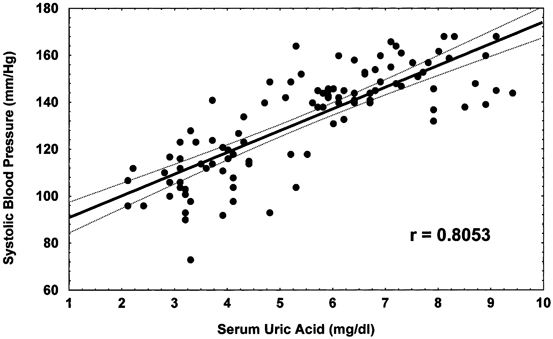
Fig. 12.3
Linear relation between serum uric acid and blood pressure. Serum uric acid is plotted against systolic BP for children with normal BP (controls) and primary hypertension. Solid and dotted lines in both panels represent the best fit and 95 % confidence intervals, respectively, and demonstrate the linear relation between uric acid concentration and systolic BP. Pearson correlation coefficients are r = 0.8053 (P = 0.000004) for systolic BP. BP blood pressure. (Reprinted from [18]. With permission from Wolters Kluwer Health)
Lowering uric acid with either allopurinol or probenecid has also been reported to reduce BP markedly in pilot studies conducted in adolescents with hypertension or prehypertension [19, 20]. In a randomized, double-blind, placebo-controlled, crossover study involving newly diagnosed adolescents with stage 1 essential hypertension and uric acid levels > 6 mg/dL, subjects were treated with allopurinol or placebo. Treatment with allopurinol resulted in normalization of BP in two-thirds of the subjects compared to one participant in the placebo group. Another recent study of prehypertensive obese adolescents reproduced similar results and provided direct evidence that uric acid increases BP in adolescents and that this effect can be mitigated by lowering uric acid. In this randomized, double-blind, placebo-controlled study, 60 children (aged 11–17) with prehypertension and uric acid 5 mg/dl were treated with either allopurinol, probenecid, or placebo for 8 weeks. Based on the clinic and 24-h ambulatory BP measurements, patients in the active treatment groups with allopurinol and probenecid experienced marked reduction in systolic BP (average 10 mmHg) and diastolic BP (average 7 mmHg). An interesting observation in this study was the effect of therapy on weight gain. Patients in the treatment arm had stable weight during the 3-month study period compared to the placebo group which gained ~ 1 kg per month. Similar observations were noted in another study which showed increased BP, weight, and metabolic syndrome in young adult men taking a high-fructose diet (which raises uric acid levels), and these effects were mitigated by allopurinol [21] . Epidemiologic studies have also shown that uric acid can predict weight gain [22]. All these striking findings strongly highlight the role of uric acid in hypertension and other features of metabolic syndrome like obesity.
The beneficial effects of lowering uric acid in adults are less marked, consistent with the findings in animal studies that once the injury has occurred, hypertension persists irrespective of uric acid levels [23].
The Link Between Uric Acid and Hypertension May Begin in the Intrauterine Environment
The importance of the intrauterine environment in influencing BP during adult life was first reported by Barker et al. who showed an inverse relationship between birth weight and systolic BP [24]. Recent studies suggest that serum uric acid may have a role in both causing low birth weights and increasing the risk for development of hypertension later on in life [25]. Elevated uric acid levels seen during normotensive pregnancy [26] or with preeclampsia predict low birth weight [27] and may have a negative effect on fetal growth and kidney development by blocking endothelial cell proliferation and function [25, 28, 29]. Studies examining children with a history of low birth weight show impaired endothelial function and increased BP and serum uric acid levels [30, 31]. While it is not known if the relationship of elevated uric acid in the mother and child reflects genetic factors, epigenetic factors, or diet, existing data clearly suggest a strong relationship of uric acid with birth weight and the risk for development of hypertension as an adult.
Proposed Mechanism for Uric Acid-Induced Hypertension
The mechanism by which serum uric acid may have a role in driving hypertension is complex . Serum uric acid reflects extracellular uric acid and is directly linked with gout, which results from extracellular deposition of urate crystals in joints and tissues. However, the vascular and renal effects of uric acid are likely mediated by intracellular uric acid levels. While an elevated serum uric acid usually translates into increased intracellular levels due to uptake in cells via organic anion transporters such as URAT1, it remains possible that alterations in urate transport mechanisms might alter this relationship.
A key aspect of the dichotomy between intracellular and extracellular uric acid is the relationship with oxidative stress. Outside the cell, uric acid appears to function as an antioxidant, and is capable of inactivating superoxide anion and hydroxyl radicals. However, inside the cell, uric acid acts as a prooxidant, and this stimulates the release of inflammatory mediators, vasoconstrictors, growth factors, and oxidants [11, 13, 29, 32–35]. The oxidative burst is mediated by an increase in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and causes mitochondrial dysfunction [36, 37] . Within the vasculature, this leads to endothelial dysfunction, arteriolopathy, impaired autoregulation, and increased systemic and glomerular hydrostatic pressure [38]. Low-grade tubular injury and renal inflammation also occur, which may also contribute to salt-sensitive hypertension through pathways involving T cell infiltration and the release of oxidants and angiotensin II [39] . An overall proposed mechanism for hypertension is shown in Fig. 12.4.
Related posts:
 Neurogenic Factors in Hypertension Associated With Chronic Kidney Disease
Dual Inhibitors: RAAS Blockers/Combination Therapies: What Do All These Trials Mean?
Genome-Wide Association Studies (Gwas) of Blood Pressure in Different Populations
Neurogenic Factors in Hypertension Associated With Chronic Kidney Disease
Dual Inhibitors: RAAS Blockers/Combination Therapies: What Do All These Trials Mean?
Genome-Wide Association Studies (Gwas) of Blood Pressure in Different Populations
 Resistant Hypertension in Patients with Chronic Kidney Disease
Resistant Hypertension in Patients with Chronic Kidney Disease
 Hypertension in Children with Chronic Kidney Disease
Hypertension in Children with Chronic Kidney Disease
 Changes in Guideline Trends and Applications in Practice: JNC 2013 and the Future
Changes in Guideline Trends and Applications in Practice: JNC 2013 and the Future
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


