In the last 10 years, anti-tumor necrosis factor (TNF)-α therapy has become a cornerstone in the management of autoimmune diseases. Clinical trial data have consistently found that infliximab, adalimumab, and recently certolizumab pegol offer therapeutic benefits to patients with inflammatory bowel diseases (Crohn’s disease and ulcerative colitis). Recent understanding on how these monoclonal antibodies evoke changes at the physiological and molecular levels have provided insights into disease pathogenesis and helped to identify new targets for future drug therapy. With increased experience in the use of these anti-TNF-α antibodies the long-term safety data, use in pregnancy have become available. This article provides an overview of the current knowledge regarding anti-TNF-α therapies for clinicians caring for patients with Crohn’s disease and ulcerative colitis.
Inflammatory bowel diseases are characterized by a dysfunctional intestinal immune system, associated with the imbalanced upregulation of helper T cells (Th), leukocyte trafficking, chemokines, and tissue repair molecules. As with some other autoimmune diseases the overproduction of tumor necrosis factor (TNF)-α, by monocytes and macrophages is prevalent in the inflamed tissues. In patients with Crohn’s disease (CD), the inflamed intestinal tissues have unusually high levels of TNF-α in the deeper layers of the lamina propria and the submucosa whereas ulcerative colitis (UC) patients have high levels of TNF-α in the subepithelium and the upper layers of the lamina propria. The persistent association of unusually high levels of proinflammatory TNF-α in the diseased tissues made this cytokine an obvious target for therapeutic intervention.
TNF-α, a 51 kDa trimer, mediates intercellular communication and plays a central role in the inflammatory response. TNF-α is found on cell surfaces or in solution following enzymatic cleavage by TNF-α converting enzyme (TACE). Transmembrane TNF-α, but not soluble TNF-α, is characterized as “bipolar” because it transmits signals as both a ligand and a receptor in cell-to-cell contact. Both membrane and soluble forms of TNF-α can bind to one of two TNF receptors and initiate the signaling cascade. The TNF signal transduction pathway is also involved in cellular metabolism, thrombosis, apoptosis, and fibrinolysis pathways, resulting in granuloma formation. In CD and UC, TNF-α is believed to affect the intestinal barrier by inducing apoptosis in the villi, inducing epithelial cells to secrete chemokines, and increase production of adhesion molecules such as E-selectin. Mucosal T cells are also triggered to increase production of interferon (IFN)-γ, enhancing the immune response.
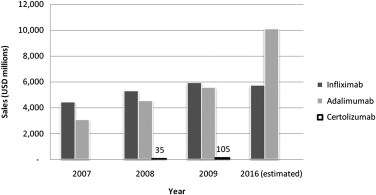
Recombinant monoclonal antibody technology was used to develop the first generation of anti-inflammatory biologic agents directed at “neutralizing” TNF-α. In 1998, the US Food and Drug Administration (FDA) approved the use of infliximab for the treatment of CD ( Table 1 ). The success of anti–TNF-α agents in CD and other autoimmune-mediated inflammatory diseases, such as rheumatoid and psoriatic arthritis, psoriasis, and ankylosing spondylitis, is evidenced by the successive development of other monoclonal antibodies, such as adalimumab and certolizumab pegol. Compared with traditional therapeutic compounds, monoclonal antibodies are less expensive to develop but more expensive to produce en masse; annual prescription costs for inflammatory bowel disease are approximately US$20,000 per patient. Nevertheless, these drugs are much needed and many have been approved for use worldwide. For example, as of January 2010, adalimumab (Humira) has been approved for use in 82 countries and used to treat 420,000 patients. By 2016, global sales forecasts expect adalimumab to be the top-selling pharmaceutical, closely followed by infliximab (Remicade) ( Fig. 1 ).
| Infliximab | Adalimumab | Certolizumab | |
|---|---|---|---|
| Crohn’s disease | Adults and children (>6 y of age) a including fistulizing disease | Adults | Adults |
| Ulcerative colitis | Adults a | ||
| Company | Centocor Ortho Biotech Inc | Abbott Laboratories | UCB |
| Brand name | Remicade | Humira | Cimzia |
| Commercial age as of April 2010 | 11.7 y | 7.3 y | 2.0 y |
a As of June 2010, approved for use by the US Food and Drug Administration but not in Canada.
In light of the recent developments and success of monoclonal antibody therapies for the treatment of inflammatory bowel diseases, the focus of this article is to characterize the clinical pharmacology of the available anti–TNF-α agents for use by clinicians on a daily basis. The 3 approved therapies for treatment of CD and UC in North America are infliximab, adalimumab, and certolizumab pegol (see Table 1 ). Trials testing the efficacy of these biologics for use for nonapproved indications are ongoing. Examples are the use of infliximab for pediatric UC or CD patients directly following intestinal surgery, although at a lower dosage (3 mg/kg instead of 5 mg/kg). Moreover, a phase IV trial has investigated the efficacy of certolizumab pegol in perianal fistulizing CD.
Of note, initial trials with etanercept for the treatment of CD proved to be ineffective and were discontinued, although it has been successful in the treatment of other immune-mediated diseases such as rheumatoid arthritis. This suggests the mechanism of action of anti–TNF-α agents for CD is not entirely related to the simple suppression of TNF-α. Etanercept is a fusion protein produced using recombinant DNA, where the DNA “construct” is engineered to link the human gene for soluble TNF receptor 2 to the gene for the Fc component of human immunoglobulin G1 (IgG1).
Anti–TNF-α monoclonal antibody structure
Infliximab (Remicade) is a chimeric mouse-human recombinant monoclonal antibody comprising a 25% variable murine Fab′ region linked by disulfide bonds to the 75% human IgG1:κ Fc constant region ( Table 2 ).
| Infliximab | Adalimumab | Certolizumab | |
|---|---|---|---|
| Half-life | 7.7–9.5 d | 10–20 d | 14 d |
| Molecular structure | Mouse-human Anti-TNF IgG1 | Fully human Anti-TNF IgG1 | Anti-TNF Fab′ fragment PEG |
| Molecular weight | ∼149,100 Da | ∼91,000 Da | |
| Mediates complement-dependent cytotoxicity | Yes | Yes | No |
| Mediates antibody-dependent cell-mediated cytotoxicity | Yes | Yes | No |
| Increases ratio of apoptotic cells | Yes | Yes | No |
| Route of administration | Intravenous infusions over 2 h a | Subcutaneous injections | Subcutaneous injections |
| Induction dose and schedule (wk) | 5 mg/kg 0, 2, and 6 | 160 mg at 0 wk 80 mg at 2 wk | 400 mg 0, 2, and 4 |
| Maintenance dose and schedule | 5 mg/kg Every 8 wk | 40 mg Every 2 wk | 400 mg Every 4 wk |
| Loss of response? | Can increase dosage to a maximum of 10 mg/kg and/or decrease infusion frequency (< every 8 wk) | Can add a single extra dose or repeat the induction schedule | |
| Pivotal RCTs in CD: Induction Maintenance | ACCENT I, SONIC ACCENT II | CLASSIC I CLASSIC II , CHARM | PRECISE 1 PRECISE 2 |
| Pivotal RCTs in UC: Induction Maintenance | ACT 1 ACT 2 |
a Infusion time can be reduced to 1 h to prevent adverse reactions.
Adalimumab (Humira) is also a recombinant monoclonal antibody like infliximab, but is fully humanized (a controversial topic), containing human-derived variable regions and a human IgG1:κ constant region.
Certolizumab pegol (Cimzia) contains the Fab′ fragment of a humanized anti-TNF monoclonal antibody. To increase the plasma half-life, the Fab′ was attached to a polyethylene glycol moiety, consisting of 2 branches of 20 kDa, at a free cysteine residue far removed from the antigen-binding site, to prevent interference.
Mechanism of action
Clinical trials involving infliximab, adalimumab and certolizumab have provided insight into how the different agents interrupt the dysfunctional immune system (see Table 2 ). The binding of transmembrane TNF-α has received much attention, as infliximab, adalimumab, and certolizumab each appears to exert different effects. In addition to their direct TNF-α inhibitory effects, some monoclonal antibodies targeting TNF-α have been implicated to have direct cytotoxicity and apoptotic action.
Each of the 3 anti-TNFs binds specifically to free and membrane-bound TNF-α, preventing it from binding to one of two possible receptors, TNF-R1 or TNF-R2. Infliximab and adalimumab do not bind TNF-β, lymphotoxin. The affinities, avidities, and complement activation of infliximab and adalimumab are very similar; certolizumab data are unavailable. The 3 antagonists are known to reduce levels of TNF-α, while infliximab and adalimumab also decrease serum interleukin (IL)-6 and acute-phase reactants, such as C-reactive protein. The clinical practice is aiming for healing. As mucosal healing as the gold standard for remission and treatment success, this is certainly achievable with infliximab, adalimumab, and certolizumab pegol. Similarly, infliximab has now been demonstrated to decrease intestinal hyperpermeability in CD patients and colitis-induced animals.
It is interesting that all 3 anti–TNF-α agents can inhibit production of the cytokine, IL-1β, in response to stimulation with lipopolysaccharide. This finding ties in well with the current tenet that inflammatory bowel disease is the result of the combination of environmental stimuli in genetically susceptible individuals with dysfunctional immune systems. Thus, the lipopolysaccharide layer of bacteria resident in the intestinal lumen can elicit an immune response leading to enhanced cytokine production by cells in the intestinal epithelial, endothelial, and submucosal layers. However, the exact mechanisms by which this can be accomplished are the focus of current studies. It should be noted that the observed restoration of the dysregulated expression of antimicrobial peptides in intestinal mucosa of CD and UC patients following infliximab therapy is a consequence of reduced inflammation; anti-TNFs do not exert a direct effect on these proteins.
Infliximab treatment efficacy has now been linked with ameliorating a potential systemic adaptation to active CD. Several cell types release neutrophil gelatinase–associated lipocalin (NGAL), a stress protein, in response to injury. Elevated levels of NGAL have been found in kidney disease as well as CD and UC. The effectiveness of a single infliximab treatment was mirrored by a 62% reduction in NGAL. Moreover, cytogenetic studies in CD patients have found high rates of sister chromatid exchanges in comparison with healthy controls, indicating chromosomal instability. Infliximab induction therapy reduces the rates of these exchanges.
The human IgG1 Fc regions in infliximab and adalimumab have a role in mediating apoptosis in cells expressing TNF-α. This was considered to be an important factor in the therapeutic success of anti-TNFs, however, the success of certolizumab pegol, which lacks this region and substantial apoptosis, suggests that apoptosis is not critical to the therapeutic success of antibodies to TNF-α. As an aside, the TNF-α antagonist etanercept, which possesses the human IgG1 Fc region, is ineffective in treating CD but has been very successful in the treatment of rheumatoid arthritis, suggesting that the mechanism of action of anti-TNFs is inflammatory disease specific.
Infliximab treatment for rheumatoid arthritis is the focus of mechanistic studies with results routinely extrapolated to CD and UC, although not all results are wholly transferable. Nevertheless, results of any research in this area further the understanding of the effect and drug activity in disease, and thus are important and merit inclusion. For example, rheumatoid arthritis patients who respond favorably to infliximab therapy experience an increase in circulating CD4 + CD25 + regulatory T cells. However, measures in CD patients before and after infliximab induction therapy found a nearly uniform level of circulating regulatory T cells and no measurable level of “defective” cells, such as CD 62L T regulatory cells, as in rheumatoid arthritis. Recently, infliximab has been demonstrated to redistribute Forkhead box protein3 T-regulatory cells (Foxp3Treg) in CD and UC patients. Specifically, peripheral blood levels of Foxp3Treg experience a sustained increase in response to infliximab therapy, in contrast to the downregulation of mucosal mRNA and protein levels of Foxp3Treg, effects seen only in patients responding to infliximab treatment.
Infliximab treatment in rheumatoid arthritis appears to inhibit the expression of IL-33R in neutrophils. In turn, neutrophils do not migrate to inflamed tissues in response to the chemotactic activity of IL-33, thereby dampening the immune response and allowing the tissues to recover. Likewise, in UC patients, mucosal IL-33 levels increase in relation to increased disease severity, while serum levels of the cleaved IL-33 are similarly elevated in both CD and UC patients. IL-33 and the IL-1 receptor-related protein, ST2, are known to be regulated by TNF-α. As IL-33 is a potent inducer of IL-5, IL-6, and IL-17 from mucosal immune cells, it is an agent that promotes and maintains tissue inflammation. Infliximab treatment in a murine inflammatory bowel disease model alters the expression of IL-33, causing circulating levels to decrease with a corresponding increase in soluble ST2.
As mentioned earlier, infliximab binding to the transmembrane TNF-α has the potential to evoke many changes in the TNF signal transduction pathway due to its bipolar capabilities. In CD, high levels of circulating IL-15 are associated with disease and inflammation. The IL-15 receptor α, produced by intestinal epithelial cells, binds to IL-15 thereby neutralizing its effects. Infliximab therapy has been shown to increase production of the IL-15 receptor, resulting in decreased circulating levels of IL-15 and elevation in the number of receptor-bound complexes. Bouchaud and colleagues propose that infliximab binding to the transmembrane TNF-α leads to reverse signaling, resulting in the dampening of the inflammatory cascade by including the release of IL-15 receptor α.
In vitro s tudies with peritoneal macrophages from the TNFαR1 knockout mouse showed significantly reduced downstream expression of inflammatory mediators (inducible nitric oxide synthase and cyclooxygenase-2) and actions (eg, phosphorylation) when exposed to lipopolysaccharides. Incubation with infliximab showed a similar concentration-dependent inhibition of inflammation. The results confirm that infliximab interference with binding of TNF-α with the TNFR1 reduces the inflammatory response when exposed to “foreign” bacterial lipopolysaccharides.
In UC, infliximab has been shown to downregulate the expression of TNF-α and IFN-γ mRNA in inflamed colonic mucosa but not in IL-10 and IL-4 mRNA. Furthermore, UC patients, homozygous for IL23R variants, were twice as likely to respond to infliximab therapy as noncarriers. As such, the therapeutic effect of infliximab binding to TNF-α is amplified by the IL23R gene.
Infliximab pharmacology has been mimicked by wormwood ( Artemsia absinthium ) extract. A clinical trial of CD patients with active disease receiving the extracts confirmed TNF-α and other interleukins suppression. This result provides another avenue of investigation for future drug development.
In rheumatoid arthritis, adalimumab alters the intracellular signal transduction by exerting effects on T-cell signal transducer and activator of transcription STAT. Specifically, adalimumab increases Th2-associated STAT6 phosphorylation and restores the Th-1-associated activation-induced STAT4 phosphorylation to normal levels seen in healthy individuals.
Mechanism of action
Clinical trials involving infliximab, adalimumab and certolizumab have provided insight into how the different agents interrupt the dysfunctional immune system (see Table 2 ). The binding of transmembrane TNF-α has received much attention, as infliximab, adalimumab, and certolizumab each appears to exert different effects. In addition to their direct TNF-α inhibitory effects, some monoclonal antibodies targeting TNF-α have been implicated to have direct cytotoxicity and apoptotic action.
Each of the 3 anti-TNFs binds specifically to free and membrane-bound TNF-α, preventing it from binding to one of two possible receptors, TNF-R1 or TNF-R2. Infliximab and adalimumab do not bind TNF-β, lymphotoxin. The affinities, avidities, and complement activation of infliximab and adalimumab are very similar; certolizumab data are unavailable. The 3 antagonists are known to reduce levels of TNF-α, while infliximab and adalimumab also decrease serum interleukin (IL)-6 and acute-phase reactants, such as C-reactive protein. The clinical practice is aiming for healing. As mucosal healing as the gold standard for remission and treatment success, this is certainly achievable with infliximab, adalimumab, and certolizumab pegol. Similarly, infliximab has now been demonstrated to decrease intestinal hyperpermeability in CD patients and colitis-induced animals.
It is interesting that all 3 anti–TNF-α agents can inhibit production of the cytokine, IL-1β, in response to stimulation with lipopolysaccharide. This finding ties in well with the current tenet that inflammatory bowel disease is the result of the combination of environmental stimuli in genetically susceptible individuals with dysfunctional immune systems. Thus, the lipopolysaccharide layer of bacteria resident in the intestinal lumen can elicit an immune response leading to enhanced cytokine production by cells in the intestinal epithelial, endothelial, and submucosal layers. However, the exact mechanisms by which this can be accomplished are the focus of current studies. It should be noted that the observed restoration of the dysregulated expression of antimicrobial peptides in intestinal mucosa of CD and UC patients following infliximab therapy is a consequence of reduced inflammation; anti-TNFs do not exert a direct effect on these proteins.
Infliximab treatment efficacy has now been linked with ameliorating a potential systemic adaptation to active CD. Several cell types release neutrophil gelatinase–associated lipocalin (NGAL), a stress protein, in response to injury. Elevated levels of NGAL have been found in kidney disease as well as CD and UC. The effectiveness of a single infliximab treatment was mirrored by a 62% reduction in NGAL. Moreover, cytogenetic studies in CD patients have found high rates of sister chromatid exchanges in comparison with healthy controls, indicating chromosomal instability. Infliximab induction therapy reduces the rates of these exchanges.
The human IgG1 Fc regions in infliximab and adalimumab have a role in mediating apoptosis in cells expressing TNF-α. This was considered to be an important factor in the therapeutic success of anti-TNFs, however, the success of certolizumab pegol, which lacks this region and substantial apoptosis, suggests that apoptosis is not critical to the therapeutic success of antibodies to TNF-α. As an aside, the TNF-α antagonist etanercept, which possesses the human IgG1 Fc region, is ineffective in treating CD but has been very successful in the treatment of rheumatoid arthritis, suggesting that the mechanism of action of anti-TNFs is inflammatory disease specific.
Infliximab treatment for rheumatoid arthritis is the focus of mechanistic studies with results routinely extrapolated to CD and UC, although not all results are wholly transferable. Nevertheless, results of any research in this area further the understanding of the effect and drug activity in disease, and thus are important and merit inclusion. For example, rheumatoid arthritis patients who respond favorably to infliximab therapy experience an increase in circulating CD4 + CD25 + regulatory T cells. However, measures in CD patients before and after infliximab induction therapy found a nearly uniform level of circulating regulatory T cells and no measurable level of “defective” cells, such as CD 62L T regulatory cells, as in rheumatoid arthritis. Recently, infliximab has been demonstrated to redistribute Forkhead box protein3 T-regulatory cells (Foxp3Treg) in CD and UC patients. Specifically, peripheral blood levels of Foxp3Treg experience a sustained increase in response to infliximab therapy, in contrast to the downregulation of mucosal mRNA and protein levels of Foxp3Treg, effects seen only in patients responding to infliximab treatment.
Infliximab treatment in rheumatoid arthritis appears to inhibit the expression of IL-33R in neutrophils. In turn, neutrophils do not migrate to inflamed tissues in response to the chemotactic activity of IL-33, thereby dampening the immune response and allowing the tissues to recover. Likewise, in UC patients, mucosal IL-33 levels increase in relation to increased disease severity, while serum levels of the cleaved IL-33 are similarly elevated in both CD and UC patients. IL-33 and the IL-1 receptor-related protein, ST2, are known to be regulated by TNF-α. As IL-33 is a potent inducer of IL-5, IL-6, and IL-17 from mucosal immune cells, it is an agent that promotes and maintains tissue inflammation. Infliximab treatment in a murine inflammatory bowel disease model alters the expression of IL-33, causing circulating levels to decrease with a corresponding increase in soluble ST2.
As mentioned earlier, infliximab binding to the transmembrane TNF-α has the potential to evoke many changes in the TNF signal transduction pathway due to its bipolar capabilities. In CD, high levels of circulating IL-15 are associated with disease and inflammation. The IL-15 receptor α, produced by intestinal epithelial cells, binds to IL-15 thereby neutralizing its effects. Infliximab therapy has been shown to increase production of the IL-15 receptor, resulting in decreased circulating levels of IL-15 and elevation in the number of receptor-bound complexes. Bouchaud and colleagues propose that infliximab binding to the transmembrane TNF-α leads to reverse signaling, resulting in the dampening of the inflammatory cascade by including the release of IL-15 receptor α.
In vitro s tudies with peritoneal macrophages from the TNFαR1 knockout mouse showed significantly reduced downstream expression of inflammatory mediators (inducible nitric oxide synthase and cyclooxygenase-2) and actions (eg, phosphorylation) when exposed to lipopolysaccharides. Incubation with infliximab showed a similar concentration-dependent inhibition of inflammation. The results confirm that infliximab interference with binding of TNF-α with the TNFR1 reduces the inflammatory response when exposed to “foreign” bacterial lipopolysaccharides.
In UC, infliximab has been shown to downregulate the expression of TNF-α and IFN-γ mRNA in inflamed colonic mucosa but not in IL-10 and IL-4 mRNA. Furthermore, UC patients, homozygous for IL23R variants, were twice as likely to respond to infliximab therapy as noncarriers. As such, the therapeutic effect of infliximab binding to TNF-α is amplified by the IL23R gene.
Infliximab pharmacology has been mimicked by wormwood ( Artemsia absinthium ) extract. A clinical trial of CD patients with active disease receiving the extracts confirmed TNF-α and other interleukins suppression. This result provides another avenue of investigation for future drug development.
In rheumatoid arthritis, adalimumab alters the intracellular signal transduction by exerting effects on T-cell signal transducer and activator of transcription STAT. Specifically, adalimumab increases Th2-associated STAT6 phosphorylation and restores the Th-1-associated activation-induced STAT4 phosphorylation to normal levels seen in healthy individuals.
Related posts:
 Nonsteroidal Antiinflammatory Drug-Related Injury to the Gastrointestinal Tract: Clinical Picture, Pathogenesis, and Prevention
Nonsteroidal Antiinflammatory Drug-Related Injury to the Gastrointestinal Tract: Clinical Picture, Pathogenesis, and Prevention
 New Pharmacologic Therapies for Gastroenteropancreatic Neuroendocrine Tumors
New Pharmacologic Therapies for Gastroenteropancreatic Neuroendocrine Tumors
 Pharmabiotic Manipulation of the Microbiota in Gastrointestinal Disorders, from Rationale to Reality
Pharmabiotic Manipulation of the Microbiota in Gastrointestinal Disorders, from Rationale to Reality
 Current Medical Treatments of Dyspepsia and Irritable Bowel Syndrome
Current Medical Treatments of Dyspepsia and Irritable Bowel Syndrome
 Clinical Pharmacology of 5-ASA Compounds in Inflammatory Bowel Disease
New Pharmacologic Therapies in Gastrointestinal Disease
Clinical Pharmacology of 5-ASA Compounds in Inflammatory Bowel Disease
New Pharmacologic Therapies in Gastrointestinal Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


