Fig. 6.1
Current pathophysiological hypotheses involved in relapsing idiopathic nephrotic syndrome (INS). Clinical and experimental observations suggest that lymphocytes and podocyte disturbances are two sides of relapsing INS. Besides T-lymphocyte abnormalities, recent evidence of B-lymphocyte depletion efficacy in sustained remission suggests that B lymphocytes may play a crucial role in INS pathogenesis. It’s currently thought that relapsing INS is caused by a circulating factor which alters podocyte function resulting in nephrotic proteinuria
This review presents the major findings about the immunopathogenesis of MCNS and discusses the potential role of c-mip (c-maf-inducing protein) in the podocyte and lymphocyte alterations observed in this disorder.
6.2 MCNS Is Associated with Profound Disturbances of Cytokine Production
It has been suggested that MCNS is the clinical manifestation of a primary disorder of T-lymphocyte function leading to cytokine release that in turn alters the glomerular filtration barrier [4, 15]. Consistent with this hypothesis, various alterations in cytokine production during MCNS have been described by many studies. Despite some inconsistencies in their findings, these studies suggest that the disease is associated with perturbations of the transcriptional machinery of immune cells [5]. Discrepancies between studies may in part result from the different immunogenetic background of the patients, the lack of standardization of sampling techniques, and the diversity of methods used to measure cytokine concentrations [5].
In a study aiming to understand the molecular mechanisms underlying T-lymphocyte dysfunction in MCNS, we examined mRNA that was differentially expressed between T lymphocytes isolated from MCNS patients in relapse and those in remission. This analysis identified 84 differentially expressed transcripts, including 12 encoding proteins of yet unknown function and 30 novel transcripts [16]. Among the 42 previously characterized transcripts, at least 18 encoded proteins closely involved in T-cell receptor (TCR)-mediated complex signaling pathways. Among the genes of which the expression was found downregulated during MCNS relapse, the interleukin (IL)-12 receptor β2 subunit is particularly interesting. This molecule is selectively induced in Th1 cells suggesting that activated T lymphocytes in MCNS patients are early driven toward the Th2 phenotype. In agreement with this hypothesis, MCNS patients often exhibit a defect in the delayed-type hypersensitivity response, suggesting that Th1-dependent cellular immunity is perturbed in these individuals [17]. Moreover, food allergens and atopy have been implicated as triggering factors in some cases of MCNS [18]. IL-13, a well-known Th2 cytokine, was found upregulated in patients undergoing nephrotic relapse but not in those in remission [19]. Interestingly, the IL-13 receptor is expressed in glomeruli from MCNS patients, and IL-13 stimulates transcellular ion transport [20]. Although IL-13 does not affect cellular permeability to macromolecules in vitro [20], transgenic rats overexpressing IL-13 develop proteinuria and MCNS-like lesions [21]. Additional evidence that MCNS is associated with T-helper differentiation has come from the isolation of c-maf (cellular (c) homolog of Avian musculoaponeurotic fibrosarcoma (MAF) proto-oncogene) by subtractive cloning [16]. This protein was initially identified as a Th2-specific transcription factor that binds to the IL-4 proximal promoter and therefore regulates IL-4 expression [22]. Valanciute et al. showed that c-maf is strongly expressed in MCNS patients; during relapse, c-maf is localized to the nucleus, whereas during remission, it shuttles to the cytoplasm [23]. Contrasting with the nuclear expression of c-maf, low levels of IL-4 were detected during relapses suggesting that the downstream target gene of c-maf is not IL-4 in this context. Recent data suggest that c-maf plays a critical role in development and maintenance of follicular T-helper cells through the production of IL-21 [24]. Interestingly, levels of both IL-4 and IL-21 are low in patients in relapse, who also exhibit a high abundance of c-maf in the nuclear compartment of CD4+ T cells ([23] and unpublished data), suggesting that the target genes of c-maf in MCNS remains to be clarified. The induction of c-maf during MCNS seems to be related to that of c-mip, an 85 kDa protein encoded by a gene initially identified in the human brain [25]. Grimbert et al. demonstrated that c-mip is strongly expressed in CD4+ T cells from MCNS patients in relapse and that its forced expression in vitro in Jurkat cells stimulates c-maf signaling pathway [26]. The predicted protein structure of c-mip includes an N-terminal region containing a pleckstrin homology domain, a middle region containing several interacting docking sites including a 14-3-3 module, a protein kinase C domain, a Src homology 3 domain similar to the p85 regulatory subunit of phosphatidylinositol 3-kinase, and a C-terminal region containing a leucine-rich repeat domain. The basal expression of c-mip transcript is very low in adult tissues including the thymus, T cells, and kidney [26]. Several studies summarized below suggest a role for c-mip in the pathogenesis of MCNS.
6.3 Perturbations in T-Lymphocyte Signal Transduction in MCNS: Potential Implication of c-mip
We and others have previously reported that MCNS is associated with high nuclear factor-κB (NF-κB) activity involving p50/RelA complexes in peripheral blood mononuclear cells from patients in relapse, with levels returning to normal during remission [27, 28]. NF-κB is a transcription factor that regulates the expression of a wide variety of genes, including cytokines and chemokines, that may play a potential role in the pathogenesis of MCNS [29]. In resting T cells, the NF-κB complex is sequestrated into the cytoplasm by inhibitor of κB (I-κB) proteins. Upon T-lymphocyte activation, I-κBα is rapidly phosphorylated and subsequently degraded by the proteasome, thus allowing the nuclear translocation of NF-κB, which is then free to activate its target genes [30]. We previously identified cross talk between c-mip and NF-κB. Indeed, we found that RelA, the master regulator of NF-κB, binds to a κB palindromic consensus sequence located in the c-mip proximal promoter and inhibits its transcriptional activation in activated cells in vitro and in vivo [31]. However, in some pathological situations, the induction of c-mip is associated with the downregulation of RelA expression. Studies involving transgenic models and in vitro analyses have shown that c-mip does not interfere with the transcriptional regulation of RelA [32]. Instead, c-mip binds RelA through its leucine-rich repeat domain and targets it to the proteasome for degradation [32, 33]. Thus, cross talk between c-mip and NF-κB seems to have functional consequences in physiological and pathological conditions but its importance is far from understood. NF-κB prevents cells from undergoing apoptosis [34], whereas c-mip facilitates the proapoptotic process by upregulating death-associated protein kinases and Bax and inhibits the antiapoptotic protein Bcl-2 and Akt [32, 35]. Cross talk between c-mip and NF-κB also influences cytokine expression. Indeed, RelA binds to the IL-4 promoter and counteracts its transcriptional activation by c-maf [23]. This mechanism, called transcriptional interference, has been poorly studied in MCNS.
6.4 Identification of c-mip as a Molecular Marker of Hodgkin Lymphoma-Associated MCNS
In patients suffering from both classical Hodgkin lymphoma (cHL) and MCNS, the course of the two diseases is remarkably similar. Notably, remission of MCNS occurs after the successful treatment of cHL, regardless of the therapeutic strategy used, suggesting that MCNS is a paraneoplastic syndrome in the context of cHL [6]. Clearly, no particular subgroup of patients with cHL, with respect to age, sex, or disease stage, seems to be at high risk of developing MCNS (except for systemic symptoms and inflammatory syndrome, which occur more frequently in cHL patients with MCNS than without MCNS) [6]. The pathogenesis of this association remains poorly understood and the underlying molecular mechanism is still unknown. We recently reported that c-mip may play a crucial role in the occurrence of MCNS in the context of cHL. Many aspects related to T-lymphocyte function have been explored in these two diseases. Th2 cytokines, in particular IL-13, seem to be involved in the pathogenesis of both diseases [21, 36], and tumoral lymphocytes, called Hodgkin and Reed-Sternberg (HRS) cells, exhibit consistent NF-κB activation [37]. We demonstrated that c-mip is selectively induced in both podocytes and HRS cells in patients with this association but not in lymphomatous tissue from cHL patients without MCNS (Fig. 6.2) [38]. We also showed that the forced expression of c-mip in HRS cell lines (which do not constitutively express c-mip) inhibits endogenous phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a transmembrane adaptor protein, and negatively regulates early proximal signaling. Furthermore, we found that the strong expression of c-mip in patients with cHL associated with MCNS may be related to a defect in Fyn (a major podocyte and lymphocyte SRC kinase) in both HRS cells and podocytes. These findings suggest that the expression of c-mip in HRS cells from lymphomatous tissues of cHL patients may be a relevant biomarker of MCNS associated with cHL. The potential role of c-mip as a negative regulator of proximal signaling in immune cells is supported by its interaction with the p85 subunit of PI3-kinase, leading to Lck inhibition [39]. We recently described the clinical, biological, and pathological spectrum of MCNS associated with non-Hodgkin lymphoid disorders [7]. MCNS occurs preferentially with neoplasms originating from B lymphocytes (94.4 % of cases). The potential role of c-mip in MCNS related to non-Hodgkin lymphoma requires further investigation, but preliminary results suggest that c-mip interferes with B-lymphocyte signaling pathways (unpublished data).
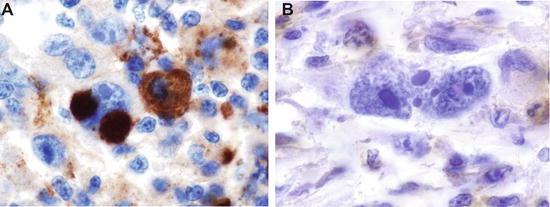
Fig. 6.2
c-mip is selectively expressed in HRS cells from patients with MCNS associated with classical Hodgkin lymphoma (cHL). MCNS is the most frequent glomerular disease associated with cHL. C-mip is selectively induced in tumoral cells (HRS cells) in patients with cHL associated MCNS but not in patients with isolated cHL suggesting its potential involvement in the pathogenesis of this association. (a) c-mip expression in one patient with MCNS and cHL. (b) c-mip expression in one patient with isolated cHL without MCNS
6.5 Do Similar Mechanisms Account for T-Lymphocyte and Podocyte Dysfunction in MCNS?
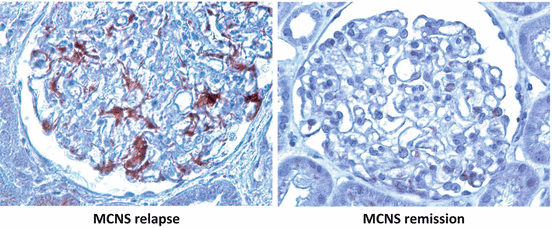
MCNS is currently considered a two-faceted disease whereby primary immune dysfunction consequently leads to podocyte injury (Fig. 6.1). Unexpectedly, we found that c-mip is upregulated in the podocytes during MCNS relapse but repressed in remission (Fig. 6.3). We thus investigated the potential role of c-mip in podocyte signaling. Nephrin is a central component of the slit diaphragm and mediates outside-in signaling in podocytes to maintain their integrity [40]. In physiological conditions, Fyn binds phosphorylates and activates nephrin signaling pathway. Functional studies in vitro in podocyte cell lines stably transfected with c-mip have shown that its overexpression is associated with alterations of podocyte proximal signaling, including the inactivation of Fyn and Akt signaling pathways and cytoskeletal disorganization [35]. These findings led us to generate a transgenic mouse model in which we used targeted transgenesis to express c-mip specifically in podocytes under the control of the nephrin promoter. These transgenic mice developed heavy proteinuria without inflammatory lesions or infiltrating cells. Furthermore, glomeruli appeared normal on light microscopy, and podocyte foot process effacement was the only morphological change observed during ultrastructural analysis. Thus, the renal phenotype observed in c-mip transgenic mice resembles closely that observed in MCNS patients. Biochemical analyses in vivo and immunomorphological studies showed that c-mip alters podocyte signaling and cytoskeleton remodeling, which is an important event in the induction of proteinuria [35]. The inactivation of nephrin and Akt has been confirmed by immunohistochemistry studies of renal biopsies from MCNS patients, emphasizing the crucial role of c-mip in podocyte dysfunction in these patients [35]. However, the robust transcriptional and posttranscriptional activation of c-mip is not restricted to MCNS, because it is also observed in primary FSGS and membranous nephropathy (MN) [32, 35, 41]. Nevertheless, c-mip is not expressed in inflammatory and proliferative glomerular diseases such as IgA nephropathy or lupus nephritis or in HIV-associated nephropathy and diabetic nephropathy [32, 35]. Strikingly, most proteins involved in TCR proximal signaling pathway (Fyn, PAG, and Csk) are constitutively expressed in human glomeruli suggesting that this regulatory loop is operational in human podocytes in addition to lymphocytes [38]. Experiments demonstrating that the inhibition of c-mip significantly improves proteinuria provide the strongest evidence for the role of c-mip in podocyte injury and subsequent proteinuria in some primary glomerular diseases including MCNS, idiopathic FSGS, and MN. In an experimental model of MN, resulting from the administration of anti-megalin antibodies (passive-type Heymann nephritis (PHN)), we found that c-mip expression in podocytes was correlated with heavy proteinuria [41]. Rats with PHN treated by cyclosporine A (CsA) display a significant decrease in the severity of proteinuria concomitant with a reduction of c-mip expression assessed by quantitative RT-PCR and immunohistochemistry. In parallel, levels of active RhoA, nephrin, and synaptopodin, which were significantly lower in PHN rats than in control animals, were restored upon CsA treatment. Moreover, Zhang et al. prevented the expression of c-mip by RNA interference in a model of podocyte damage after LPS administration and found that the severity of proteinuria was 70 % lower in mice deficient for c-mip than in control mice [35]. These experimental results in several models of podocyte injury suggest that targeting c-mip with specific inhibitors taken up by podocytes may be a promising future therapeutic approach for some glomerular diseases associated with the expression of c-mip in podocytes.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>
 History of Research on Pathogenesis of Idiopathic Nephrotic Syndrome
Regulatory T Cells and Oxidative Stress in Minimal Change Nephropathy
Cytokines as Active Factors in Minimal Change Nephrotic Syndrome
Podocytes as a Direct Target of Drugs Used in Idiopathic Nephrotic Syndrome
Co-stimulatory Molecule CD80 (B7.1) in MCNS
History of Research on Pathogenesis of Idiopathic Nephrotic Syndrome
Regulatory T Cells and Oxidative Stress in Minimal Change Nephropathy
Cytokines as Active Factors in Minimal Change Nephrotic Syndrome
Podocytes as a Direct Target of Drugs Used in Idiopathic Nephrotic Syndrome
Co-stimulatory Molecule CD80 (B7.1) in MCNS
 Cationic Bovine Serum Albumin as Cause of Membranous Nephropathy: From Mice to Men
Cationic Bovine Serum Albumin as Cause of Membranous Nephropathy: From Mice to Men




Only gold members can continue reading. Log In or Register to continue
Related posts:
History of Research on Pathogenesis of Idiopathic Nephrotic Syndrome
Regulatory T Cells and Oxidative Stress in Minimal Change Nephropathy
Cytokines as Active Factors in Minimal Change Nephrotic Syndrome
Podocytes as a Direct Target of Drugs Used in Idiopathic Nephrotic Syndrome
Co-stimulatory Molecule CD80 (B7.1) in MCNS
Cationic Bovine Serum Albumin as Cause of Membranous Nephropathy: From Mice to Men
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
