The Patient with Hypokalemia or Hyperkalemia
Jie Tang
Stuart L. Linas
Potassium is the most abundant cation in the human body. It regulates intracellular enzyme function and helps determine neuromuscular and cardiovascular tissue excitability. Over 98% of total body potassium is located in the intracellular fluid (ICF; primarily in muscles), less than 2% in the extracellular fluid (ECF). The ratio of extracellular potassium to intracellular potassium determines membrane potential. The acuity of changes in serum potassium concentration and membrane potential determines the severity of clinical symptoms and underlies the clinical findings caused by disorders of potassium metabolism.
I. OVERVIEW OF POTASSIUM PHYSIOLOGY.
The typical Western diet contains 40 to 120 mEq of potassium a day. Tight control of the serum potassium between 3.5 and 5.5 mEq/day is primarily accomplished by the kidney where secretion varies between 40 and 120 mEq/day. Potassium losses in stool and sweat are small (5 to 10 mEq). In addition, the interplay of several hormonal systems and the internal acid—base environment contribute to the exchange of potassium between the ECF and ICF, which helps keep the serum potassium concentration tightly controlled. Although total body potassium declines with aging, and the rate of decline appears to be influenced by sex and race, the clinical significance of these observations is not clear.
A. Internal Balance. Under certain physiological conditions, potassium is rapidly redistributed between the intracellular and extracellular compartments. Several hormones and physiological factors interact to regulate the transcellular movement of potassium.
1. Insulin. High serum potassium increases insulin levels. The binding of the insulin hormone to insulin receptors causes a hyperpolarization of cell membranes that facilitates potassium uptake in liver, fat, cardiac, and skeletal muscle. Insulin also activates Na—K—adenosine triphosphatase (ATPase) pumps and causes the cellular uptake of potassium.
2. Catecholamines. Activation of the β2-adrenoreceptor results in cellular potassium uptake in the liver and muscle. In addition to the activation of inwardly directed Na+-K+-Cl2– (NKCC) cotransporters, the effect is also transduced by cyclic adenosine monophosphate (cAMP) activation of Na—K—ATPase pumps, causing an influx of potassium in exchange for sodium. Therapeutic agents such as theophylline potentiate β2-adrenoreceptor—mediated potassium uptake by inhibiting the degradation of cAMP.
3. Acid—Base. Inorganic acidosis (e.g., hydrochloric acid) facilitates potassium movement from ICF to ECF. Protons enter cells, whereas impermeant inorganic ions do not. The resulting increases in ICF positive charge favor the outward movement of potassium. Because organic ions (lactate, ketoacids) are less restricted from entering cells, increases in serum potassium may not occur in organic acidosis.
4. Tonicity. Hyperglycemia causes potassium-rich fluid to leave the cell, thereby increasing ECF potassium. Under most conditions, increases in insulin modulate and reverse the effect of increased extracellular tonicity. However, when insulin cannot be increased (e.g., type 1 diabetes mellitus) or hyperglycemia occurs rapidly (as with the administration of 50% glucose), hyperkalemia occurs. Rapid infusions of mannitol also may cause hyperkalemia.
B. External Balance
1. Kidney
Urinary potassium excretion is the result of a difference between the potassium secreted and potassium reabsorbed in the distal nephron. Potassium is freely filtered at the glomerulus. More than 50% of filtered potassium is reabsorbed in the proximal convoluted tubule through paracellular pathways. In the descending limb of Henle’s loop, especially in deep nephrons, potassium concentration increases. In the medullary thick ascending limb of the loop of Henle, the Na—K—2Cl—cotransporter leads to the reabsorption of potassium. When the tubular fluid reaches the early distal convoluted tubule, only 10% to 15% of filtered potassium remains. Potassium is secreted by the principal cells of the connecting tubule and cortical collecting duct. Potassium is reabsorbed in the outer medullary collecting duct, an effect mediated by intercalated cells. A fall in glomerular filtration rate (GFR) is not generally associated with decreased potassium excretion and hyperkalemia until the GFR is less than 20 mL/min. It is due to an adaptive increase in potassium excretion in the remaining functioning nephrons. The major factors regulating potassium excretion follow.
a. Distal Nephron Flow Rate and Sodium Delivery. Under normal conditions, sodium delivered to the cortical collecting tubule is reabsorbed through amiloride-sensitive epithelial sodium channels (ENaCs) in the principal cells. The resulting negative potential in the tubular lumen results in increased potassium excretion through apical potassium channels [renal outer medullary potassium (ROMK) channel]. This system requires sodium delivery to the distal tubule. In addition, increases in tubular flow rate help maintain a low urinary potassium concentration, which favors the movement of potassium from cells into tubular fluid.
b. Mineralocorticoids. Aldosterone is the major mineralocorticoid; it increases potassium secretion into the tubular fluid by the following:
i. Increasing the number and activity of apical amiloride-sensitive ENaCs in the connecting tubule and cortical collecting duct in the distal tubule. This increases sodium reabsorption, thereby creating a negative lumen and driving force for potassium excretion into the tubular lumen.
ii. Increasing basolateral Na—K—ATPase activity.
c. Increases or decreases in dietary potassium increase or decrease urinary potassium, respectively. Renal adaptation to high potassium intake is mediated by a potassium-induced increase in aldosterone secretion and by an increase in distal nephron Na—K—ATPase activity. In response to potassium restriction, mineralocorticoid activity decreases, thereby causing a decline in potassium secretion.
d. Increases in relatively nonresorbable anions (e.g., bicarbonate, penicillin) trap secreted potassium in the tubular lumen and limit potassium reabsorption in the medullary collecting duct. The resulting renal potassium losses may lead to severe potassium depletion.
e. WNK kinases are a recently identified series of enzymes found to regulate potassium excretion. WNK4 decreases activity of the NaCl transporter in the distal tubules and decreases the number of potassium channels in the cortical collecting tubule. The net effect of WNK4 is to cause potassium retention.
2. Extrarenal
a. Gastrointestinal Tract
At a lower potassium intake (<55 mmol/day), there appears to be an inverse relationship between renal and gastrointestinal (GI) potassium excretions. However, at a higher intake, both renal and GI potassium excretions increase. It indicates that the GI tract may play a significant role in handling the extra potassium load, especially when the kidney function is compromised. The relative contribution of GI potassium excretion may be different in people with different racial background.
b. Others
Both salivary and sweat glands are involved in potassium excretion regulated by aldosterone. However, its clinical significance in potassium homeostasis is not clear.
II. HYPOKALEMIA
A. Diagnosis. The initial approach to hypokalemia is to determine whether it is spurious, secondary to a shift of potassium from the extracellular to intracellular compartments, or a result of a true decrease in total body potassium (Fig. 3-1).
Spurious hypokalemia occurs in the setting of extreme leukocytosis (in vitro white blood cells uptake potassium in the test tube) and is not associated with changes in either internal or external potassium balance.
Potassium shifts into cells may occur acutely in conditions associated with increases in endogenous insulin or catecholamines. For example, catecholamine release associated with shortness of breath (asthma, chronic obstructive pulmonary disease exacerbations, heart failure, and chest pain syndromes including myocardial infarction or angina) or catecholamine release from certain drug withdrawals (alcohol, narcotics, or barbiturates) shifts potassium into cells, thereby decreasing the serum potassium concentration. Hypokalemia may also be caused by insulin administration (correction of diabetic ketoacidosis, postresuscitation for hyperkalemia) or β2-adrenoreceptor agonist (β2-agonists, theophylline). Other common causes of decreases in serum potassium without decreases in total body potassium include hypokalemic periodic paralysis (familial
and hyperthyroid types), treatment of megaloblastic anemias, and refeeding syndromes (probably insulin mediated). The refeeding syndrome, in which severely malnourished patients are begun on nasogastric feeding, is also seen in older adults in whom the clinical manifestations of malnutrition are less clinically apparent.
and hyperthyroid types), treatment of megaloblastic anemias, and refeeding syndromes (probably insulin mediated). The refeeding syndrome, in which severely malnourished patients are begun on nasogastric feeding, is also seen in older adults in whom the clinical manifestations of malnutrition are less clinically apparent.
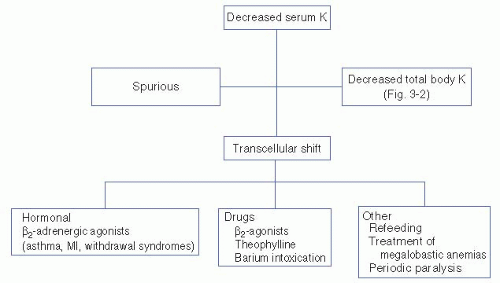 Figure 3-1. Diagnostic approach to hypokalemia. (K, potassium; MI, myocardial infarction.) |
Decreases in total body potassium (Fig. 3-2) are caused by either inadequate potassium intake or by excessive renal or extrarenal potassium losses. The measurement of urinary potassium excretion (by 24-hour measurements or “spot” potassium concentrations) is used to distinguish renal versus extrarenal potassium loss. Urinary potassium concentrations less than 20 mEq/L suggest poor potassium intake and/or extrarenal potassium loss. Serum acid—base status is helpful in evaluating hypokalemia with low urinary potassium excretion. Metabolic acidosis may suggest lower GI losses (diarrhea of any cause, e.g., infectious, toxic, and laxative abuse). A normal serum pH is less helpful because hypokalemia can be secondary to both decreases in intake and GI losses. Metabolic alkalosis with urinary potassium of less than 20 mEq/L, although rare, is associated with laxative abuse, villous adenoma, or congenital chloride-losing diarrhea.
Hypokalemia with a urinary potassium excretion of greater than 20 mEq/L suggests renal potassium wasting. The serum pH again is helpful to further evaluate etiologies. Metabolic acidosis suggests renal tubular acidosis (type 1 or type 2), diabetic ketoacidosis (osmotic diuresis), ureterosigmoidostomy, or carbonic anhydrase inhibitor use. More commonly, renal potassium losses are associated with metabolic alkalosis. In this clinical setting, the urinary chloride concentration is helpful. A low urinary chloride concentration (less than 20 mEq/L) suggests upper GI potassium losses, recent (but not current) diuretic use, or a posthypercapnic syndrome. Hypokalemia with a high urinary chloride concentration is further distinguished on the basis of the presence
or absence of hypertension. In normotensive individuals, hypokalemia with metabolic alkalosis and a high urinary chloride occurs with diuretic use (loop or distal convoluted tubule—acting diuretics), in Bartter’s and Gitelman’s syndrome, and with severe decreases in total body magnesium or potassium. Hypokalemia with renal potassium wasting, renal chloride wasting, and hypertension is further evaluated by urinary aldosterone concentrations. An elevated aldosterone level suggests either primary aldosteronism (adenoma, hyperplasia, glucocorticoid remedial) or secondary aldosteronism (renovascular or accelerated hypertension, diuretic use, renin-secreting tumor). Conversely, normal aldosterone levels with increases in serum cortisol suggest Cushing’s syndrome or exogenous steroid use. Normal cortisol and aldosterone levels indicate Liddle’s syndrome (caused by increases in the activity of the cortical collecting tubule sodium channel) or apparent mineralocorticoid excess syndrome [decreases in 11-β-hydroxysteroid dehydrogenase activity in kidney tissue (congenital, licorice ingestion) causing the mineralocorticoid receptor to respond to glucocorticoid]. Increases in urinary potassium excretion without a significant acid—base disorder are seen during the recovery phase of acute tubular necrosis, postobstructive diuresis, and magnesium depletion associated with drugs such as aminoglycosides and cisplatinum, or in myelomonocytic leukemia (secondary to lysozymuria).
or absence of hypertension. In normotensive individuals, hypokalemia with metabolic alkalosis and a high urinary chloride occurs with diuretic use (loop or distal convoluted tubule—acting diuretics), in Bartter’s and Gitelman’s syndrome, and with severe decreases in total body magnesium or potassium. Hypokalemia with renal potassium wasting, renal chloride wasting, and hypertension is further evaluated by urinary aldosterone concentrations. An elevated aldosterone level suggests either primary aldosteronism (adenoma, hyperplasia, glucocorticoid remedial) or secondary aldosteronism (renovascular or accelerated hypertension, diuretic use, renin-secreting tumor). Conversely, normal aldosterone levels with increases in serum cortisol suggest Cushing’s syndrome or exogenous steroid use. Normal cortisol and aldosterone levels indicate Liddle’s syndrome (caused by increases in the activity of the cortical collecting tubule sodium channel) or apparent mineralocorticoid excess syndrome [decreases in 11-β-hydroxysteroid dehydrogenase activity in kidney tissue (congenital, licorice ingestion) causing the mineralocorticoid receptor to respond to glucocorticoid]. Increases in urinary potassium excretion without a significant acid—base disorder are seen during the recovery phase of acute tubular necrosis, postobstructive diuresis, and magnesium depletion associated with drugs such as aminoglycosides and cisplatinum, or in myelomonocytic leukemia (secondary to lysozymuria).
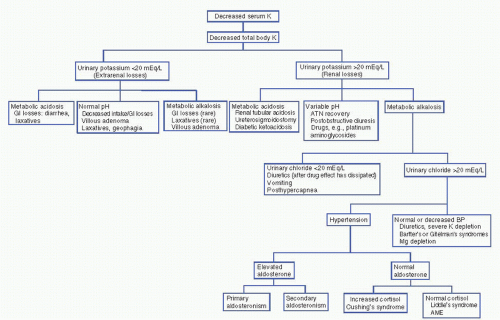 Figure 3-2. Diagnostic approach to hypokalemia. (AME, aseptic meningoencephalitis; ATN, acute tubular necroses; BP, blood pressure; GI, gastrointestinal; K, potassium; Mg, magnesium.) |
Related posts:
 The Edematous Patient: Cardiac Failure, Cirrhosis, and Nephrotic Syndrome
The Edematous Patient: Cardiac Failure, Cirrhosis, and Nephrotic Syndrome
 The Patient with Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
The Patient with Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
 The Patient with Glomerular Disease or Vasculitis
The Patient with Glomerular Disease or Vasculitis
 The Patient with Acute Kidney Injury
The Patient with Acute Kidney Injury
 The Patient with Kidney Disease and Hypertension in Pregnancy
The Patient with Kidney Disease and Hypertension in Pregnancy
 The Patient with Hypertension
The Patient with Hypertension
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


