The Patient with Disorders of Serum Calcium and Phosphorus
Jeffrey G. Penfield
Robert F. Reilly
DISORDERS OF SERUM CALCIUM
Most calcium in the body is in the form of hydroxyapatite in bone (99%). Although a small fraction of total body calcium is contained in the extracellular fluid (ECF), only the concentration of ionized calcium in the ECF is physiologically active and regulated. Approximately 60% of calcium in the ECF is ultrafilterable and exists either free in solution as ionized calcium (50%) or complexed to anions such as citrate, phosphate, sulfate, and bicarbonate (10%). The remaining 40% is bound to proteins (primarily albumin). Serum or plasma calcium concentration is measured as either total or ionized calcium. Total calcium concentration is measured with a colorimetric assay and includes ionized, complexed, and bound calcium. Ionized calcium concentration is measured with a calcium-specific electrode and represents physiologically regulated calcium. Both total and ionized calcium can be expressed in conventional units of mg per dL or mEq per L or in International System (SI) of units of mmol per L. SI units (mmol per L) can be converted to mg per dL by multiplying by 4. Measuring total calcium levels is inexpensive and convenient. A determination of the ionized calcium concentration requires that the sample be placed on ice and measured within 2 hours making it difficult for routine use, especially in the outpatient setting.
Figure 5-1 illustrates the calcium fluxes between ECF, intestine, kidney, and bone. Net intestinal calcium absorption amounts to approximately 200 mg of the normal dietary intake of 800 to 1,000 mg. In the steady state, this net intestinal absorption is matched by urinary excretion. As a result, 10,600 mg of the approximately 10,800 mg (98%) of calcium that is filtered by the glomerulus daily is reabsorbed by the renal tubules.
I. CALCIUM REGULATION.
Plasma-ionized calcium is regulated through a complex and coordinated interplay of parathyroid hormone (PTH) and 1,25(OH)2 vitamin D3 (calcitriol) in the intestine, bone, and kidney. The parathyroid gland senses ECF-ionized calcium concentration through a calcium-sensing receptor. High concentrations of ECF calcium stimulate the receptor and activate second messenger pathways that, in turn, inhibit PTH release. Low ECF calcium concentration stimulates PTH secretion and production and increases parathyroid gland mass. The parathyroid gland responds quickly (within minutes) to alterations in ionized calcium concentration. An inverse sigmoid relationship exists between ECF calcium concentration and PTH secretion, with a nonsuppressible component present even at high plasma calcium concentration. The amount of hormone stored
is enough to support basal secretion for 6 hours and stimulated secretion for 2 hours.
is enough to support basal secretion for 6 hours and stimulated secretion for 2 hours.
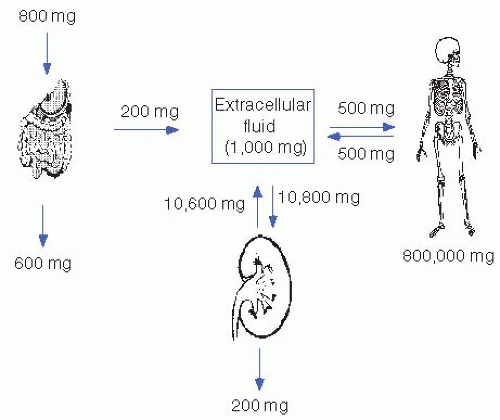 Figure 5-1. Calcium homeostasis. |
In bone, PTH in the presence of permissive amounts of calcitriol stimulates reabsorption by increasing osteoclast number and activity. In intestine, PTH enhances calcium and phosphate absorption indirectly by promoting the formation of calcitriol. In kidney, PTH augments distal tubular calcium reabsorption, stimulates calcitriol formation in the proximal tubule, and decreases proximal tubular phosphate and bicarbonate reabsorption.
Calcitriol is produced in the proximal tubule through 1α-hydroxylation of 25(OH) vitamin D3 (calcidiol). The calcitriol biosynthetic pathway is illustrated in Figure 5-2. Principal stimulators of 1α-hydroxylase are PTH and hypophosphatemia. The major function of calcitriol is to enhance calcium and phosphate availability for new bone formation and prevention of symptomatic hypocalcemia and hypophosphatemia. In intestine and kidney, calcitriol increases production of calcium-binding proteins (calbindins) that aid in transcellular calcium movement. In bone, calcitriol potentiates PTH actions, stimulates osteoclastic reabsorption, and induces differentiation of monocytes into osteoclasts.
In parathyroid gland, calcitriol binds to its receptor, leading to a decrease in PTH production. The PTH gene promoter contains regions that bind the calcitriol receptor. Binding results in a dramatic decrease in PTH expression. Calcitriol is the most potent suppressor of PTH gene transcription.
II. HYPERCALCEMIA
A. Etiology. Three basic pathophysiologic mechanisms contribute to hypercalcemia: increased calcium absorption from the gastrointestinal tract, decreased renal calcium excretion, and increased bone calcium resorption. The most common etiologies of hypercalcemia are listed in Table 5-1.
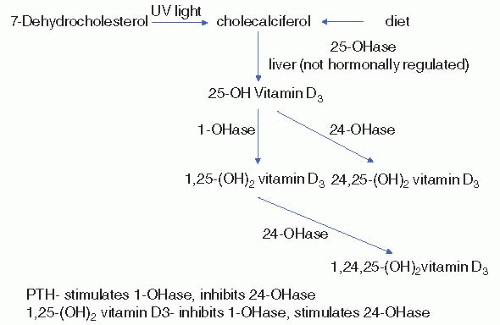 Figure 5-2. Vitamin D metabolism. (PTH, parathyroid hormone; UV, ultraviolet.) |
Table 5-1. Etiologies of Hypercalcemia | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
1. Increased calcium absorption from the gastrointestinal tract plays a primary role in the hypercalcemia of the milk-alkali syndrome, vitamin D intoxication, and granulomatous disorders.
a. Milk-alkali syndrome is the result of ingestion of excess calcium and alkali. In the past, peptic ulcer disease was treated with milk and sodium bicarbonate. This calcium and alkali source used to be the most common cause of the milk-alkali syndrome. This regimen was replaced with histamine antagonists and proton pump inhibitors so that milk and sodium bicarbonate are now rare causes of this syndrome. Currently, this syndrome most often occurs in elderly women consuming excess calcium carbonate or calcium citrate for the treatment of osteoporosis. As a result, many now refer to this as the calcium-alkali syndrome rather than the milk-alkali syndrome. Alkalosis decreases renal calcium excretion and the resultant hypercalcemia, nephrocalcinosis, and subsequent renal dysfunction prevent correction of the alkalosis. Many of these patients are also receiving vitamin D supplements that increase intestinal calcium absorption. Patients present with the classic triad of hypercalcemia, metabolic alkalosis, and elevated serum creatinine concentration. Treatment is volume replacement with normal saline and avoidance of calcium and alkali supplements. The kidney injury may persist and result in chronic kidney disease (CKD). Bisphosphonates should be avoided because these agents prevent bone calcium release, which is not a contributing factor in this syndrome and can also result in hypocalcemia. Treatment of hypercalcemia in these patients is often complicated by hypocalcemia resulting from sustained PTH suppression.
b. Hypercalcemia in CKD is uncommon, except in patients treated with calcium and vitamin D supplements. This disorder and the milkalkali syndrome illustrate the important concept that hypercalcemia from excessive dietary calcium ingestion alone does not occur in the absence of renal impairment.
c. Vitamin D intoxication also results in hypercalcemia. Calcium is absorbed primarily in the small intestine, and this process is stimulated by calcitriol.
d. Hypercalcemia may also be secondary to granulomatous disorders, such as sarcoidosis. Activated macrophages produce calcitriol, which leads to increased intestinal absorption of dietary calcium. Hypercalciuria is seen more commonly than hypercalcemia. Lymphomas occasionally cause hypercalcemia through the same mechanism.
2. Increased calcium resorption from bone plays a primary role in hypercalcemia resulting from primary, secondary, and tertiary hyperparathyroidism, malignancy, hyperthyroidism, immobilization, Paget’s disease, and vitamin A toxicity.
a. Hyperparathyroidism
i. Primary—
Primary hyperparathyroidism is a common cause of hypercalcemia. The estimated incidence ranges from 0.4 to 26 per 100,000 patient years. The incidence declined from the 1970s to 1995 but might now be increasing again. In a large multiracial US study, the
highest incidence was in African Americans followed by whites. Women are more than twice as likely as men to develop primary hyperparathyroidism and the incidence increases with advancing age. The underlying pathology is most often a solitary adenoma (80%). Among the remainder, 15% to 20% have diffuse hyperplasia, and approximately one half of these have a familial syndrome [multiple endocrine neoplasia (MEN) type 1, associated with pituitary adenomas and islet cell tumors, or MEN type II, associated with medullary carcinoma of the thyroid and pheochromocytoma]. Multiple adenomas are uncommon, and parathyroid carcinoma is rare (occurring in less than 1%). Hypercalcemia results from increased calcium resorption from bone, increased intestinal calcium absorption mediated by calcitriol, and increased distal tubular renal calcium reabsorption. In primary hyperparathyroidism, hypercalcemia is often mild, asymptomatic, and identified on routine blood chemistries in the outpatient setting.
highest incidence was in African Americans followed by whites. Women are more than twice as likely as men to develop primary hyperparathyroidism and the incidence increases with advancing age. The underlying pathology is most often a solitary adenoma (80%). Among the remainder, 15% to 20% have diffuse hyperplasia, and approximately one half of these have a familial syndrome [multiple endocrine neoplasia (MEN) type 1, associated with pituitary adenomas and islet cell tumors, or MEN type II, associated with medullary carcinoma of the thyroid and pheochromocytoma]. Multiple adenomas are uncommon, and parathyroid carcinoma is rare (occurring in less than 1%). Hypercalcemia results from increased calcium resorption from bone, increased intestinal calcium absorption mediated by calcitriol, and increased distal tubular renal calcium reabsorption. In primary hyperparathyroidism, hypercalcemia is often mild, asymptomatic, and identified on routine blood chemistries in the outpatient setting.
ii. Secondary—
Secondary hyperparathyroidism is seen in CKD patients or patients with end-stage renal disease on dialysis. Uremia causes PTH resistance and requires a higher PTH level than normal. Decreased production of 1,25(OH)2 vitamin D3 by the kidney results in less PTH suppression, as well as hypocalcemia that increases the half-life of PTH mRNA. Reduced phosphorus excretion by the kidneys results in hyperphosphatemia that also increases the half-life of PTH mRNA.
iii. Tertiary—
Tertiary hyperparathyroidism is a result of prolonged stimulation of the parathyroid gland from secondary hyperparathyroidism in end-stage renal disease. The patient will have hypercalcemia instead of hypocalcemia as a result of parathyroid gland hyperplasia. It can also be seen after renal transplantation when plasma phosphorus concentration, vitamin D metabolism, and renal function improve, but PTH secretion remains high secondary to increased parathyroid mass. In most patients, PTH levels drop and hypercalcemia resolves during the first year following transplantation.
b. Malignancy is also a common cause of hypercalcemia. Hypercalcemia of malignancy results from several pathophysiologic mechanisms: overproduction of PTH-related peptide (PTHrP), local bone reabsorption around sites of tumor infiltration (mediated through a variety of cytokines and osteolytic prostaglandins), and calcitriol production (e.g., with lymphomas). Patients with squamous cell lung cancer, breast cancer, multiple myeloma, and renal cell carcinoma are at highest risk. Hypercalcemia due to tumoral PTHrP production is often referred to as humoral hypercalcemia of malignancy (HHM). PTHrP has 70% amino acid identity to the first 13 amino acids of PTH and binds to the PTH receptor. It normally functions as a regulator of chondrocyte growth and differentiation in developing long bones; calcium mobilization from bones and into breast milk during lactation; calcium transport across the placenta to the developing fetus; and uterine blood flow. It is usually
produced in the placenta during pregnancy and by mammary glands during lactation. In certain cancers, the gene for PTHrP is inappropriately activated. HHM often presents with severe hypercalcemia (calcium concentration greater than 14 mg/dL) in a patient with either a known history of malignancy or evidence of malignancy at initial presentation. PTHrP is immunologically distinct from PTH and is not detected by standard PTH assays, but specific assays for PTHrP are commercially available. The normal range for PTHrP is less than 2.0 pmol/L because in normal health its functions are autocrine or paracrine and higher circulating levels are not required. Median survival from the onset of hypercalcemia with HHM is only 3 months. Squamous cell tumors, renal cell carcinomas, and most breast neoplasms produce PTHrP. The diagnoses of primary hyperparathyroidism and malignancy are not mutually exclusive. An increased incidence of primary hyperparathyroidism was reported in patients with malignancy.
produced in the placenta during pregnancy and by mammary glands during lactation. In certain cancers, the gene for PTHrP is inappropriately activated. HHM often presents with severe hypercalcemia (calcium concentration greater than 14 mg/dL) in a patient with either a known history of malignancy or evidence of malignancy at initial presentation. PTHrP is immunologically distinct from PTH and is not detected by standard PTH assays, but specific assays for PTHrP are commercially available. The normal range for PTHrP is less than 2.0 pmol/L because in normal health its functions are autocrine or paracrine and higher circulating levels are not required. Median survival from the onset of hypercalcemia with HHM is only 3 months. Squamous cell tumors, renal cell carcinomas, and most breast neoplasms produce PTHrP. The diagnoses of primary hyperparathyroidism and malignancy are not mutually exclusive. An increased incidence of primary hyperparathyroidism was reported in patients with malignancy.
Multiple myeloma is associated with hypercalcemia and localized osteolytic skeletal lesions. Approximately 30% of patients with myeloma experience hypercalcemia at some time during the course of their disease. Bone destruction occurs as a consequence of interleukin-6, interleukin-1, and tumor necrosis factor-beta release by malignant plasma cells. Bony lesions demonstrate a marked increase in osteoclastic resorption without manifestations of increased bone formation, in contrast to metastatic lesions of breast and prostate cancer, which generally show some increase in bone formation and radionuclide uptake at sites of increased osteoblastic activity. Because of excessive bone resorption, multiple myeloma can cause severe osteoporosis in addition to hypercalcemia. Bisphosphonates are frequently used to treat these complications. Bisphosphonates can cause acute kidney injury if given at high doses for a prolonged period of time. This risk is higher if the patient has CKD. Unfortunately, patients with multiple myeloma frequently develop CKD and standard- or low-dose bisphosphonates might not be as effective in treating the hypercalcemia and osteoporosis.
c. Hyperthyroidism results in mild hypercalcemia in 10% to 20% of patients as a result of increased bone turnover. There is also an increased prevalence of hyperparathyroidism in patients with hyperthyroidism.
d. Paget’s disease is a disease of bone with focal areas of disrupted bone turnover, disorganized and structurally weak bone, and increased vascularity. There are both hereditary and environmental factors that cause Paget’s disease. The most common mode of inheritance is autosomal dominant. Immobilization with Paget’s disease can cause hypercalcemia, although this is more likely in children. In adults, hypercalciuria is more common than hypercalcemia.
e. Miscellaneous causes of hypercalcemia include lithium use (mild; interferes with the calcium-sensing receptor); thiazide diuretic use (occult primary hyperparathyroidism should be suspected); pheochromocytoma; primary adrenal insufficiency; and a rare genetic disorder, familial hypocalciuric hypercalcemia (FHH).
FHH is an autosomal dominant disorder most commonly caused by a heterozygous mutation in the calcium-sensing receptor. It is rare with a prevalence of 1 in 78,000 in one study. Recently, mutations in two additional genes were reported to cause FHH, the G-protein subunit α11 and the S1 subunit of adaptor protein 2. The syndrome presents with mild hypercalcemia early in life, hypocalciuria, and a normal or slightly increased PTH concentration in the absence of signs or symptoms of hypercalcemia. As a result of the mutations, the calcium-sensing receptor is less sensitive to plasma calcium concentration, and a higher than normal calcium concentration is required to suppress PTH. One should be aware of FHH, because this condition is often misdiagnosed as primary hyperparathyroidism, and patients may be inappropriately subjected to neck exploration. FHH may account for a small percentage of patients who undergo surgery for primary hyperparathyroidism in whom no adenoma is found.
B. Signs and symptoms of hypercalcemia are related to the severity and rate of rise in plasma-ionized calcium concentration. Mild hypercalcemia is generally asymptomatic and often incidentally discovered on routine blood chemistries, as is the case in many patients with primary hyperparathyroidism. In contrast, severe hypercalcemia is often associated with neurologic and gastrointestinal symptoms. The patient may present with a wide range of central nervous system symptoms, from mild mental status changes to stupor and coma. Gastrointestinal symptoms include constipation, anorexia, nausea, and vomiting. Abdominal pain may result from hypercalcemia-induced peptic ulcer disease or pancreatitis. Hypercalcemia results in polyuria and secondary polydipsia can lead to hypernatremia, ECF volume contraction, a reduction in the glomerular filtration rate (GFR), and an elevation in blood urea nitrogen (BUN) and creatinine concentrations. Hypercalcemia also potentiates the cardiac effects of digitalis toxicity.
C. Diagnosis. The most common causes of hypercalcemia are primary hyperparathyroidism and malignancy. These two disorders make up more than 90% of all cases. Initial evaluation includes a history and physical examination. Use of calcium supplements, antacids, vitamin preparations, and over-the-counter medications should be determined. A chest x-ray should be obtained to rule out pulmonary malignancies and granulomatous disorders.
1. Initial laboratory examination includes measurement of electrolytes, BUN, creatinine, phosphorus, serum protein electrophoresis, and a 24-hour urine for calcium and creatinine for calculation of the calcium to creatinine clearance ratio. The presence of a high serum chloride concentration and a low serum phosphorus concentration in a ratio greater than 33:1 is suggestive of primary hyperparathyroidism resulting from the effect of PTH decreasing proximal tubular phosphate reabsorption. A low serum chloride concentration, a high serum bicarbonate concentration, and elevated BUN and creatinine concentrations are characteristic of milk-alkali syndrome. A monoclonal spike on either serum protein electrophoresis or urine protein electrophoresis is suggestive of multiple myeloma. A low serum phosphorus concentration is found in primary hyperparathyroidism and HHM. The FE of calcium is low in hypercalcemia caused by the milk-alkali syndrome, thiazide diuretic use,
or FHH. The optimal calcium to creatinine clearance ratio for separation of FHH from primary hyperparathyroidism appears to be a value of 0.0115. This yields a sensitivity of 80% and a specificity of 88%. It can be seen that even at this cutoff there is still overlap between FHH and primary hyperparathyroidism, especially in those with primary hyperparathyroidism that are also vitamin D deficient. The formula for the calcium to creatinine clearance ratio measured in a 24-hour urine is
or FHH. The optimal calcium to creatinine clearance ratio for separation of FHH from primary hyperparathyroidism appears to be a value of 0.0115. This yields a sensitivity of 80% and a specificity of 88%. It can be seen that even at this cutoff there is still overlap between FHH and primary hyperparathyroidism, especially in those with primary hyperparathyroidism that are also vitamin D deficient. The formula for the calcium to creatinine clearance ratio measured in a 24-hour urine is
All units are in mg per dL. Creatinine is abbreviated as Cr.
As a general rule, primary hyperparathyroidism is the etiology in asymptomatic outpatients with a serum calcium concentration ≤11 mg/dL, whereas malignancy is often the cause in symptomatic patients with an abrupt disease onset and serum calcium concentration ≥14 mg/dL.
2. Intact PTH concentration is obtained after the initial evaluation is completed. The most common cause of an elevated PTH concentration is primary hyperparathyroidism, although an elevated PTH concentration may also be seen with lithium use and in 15% to 20% of patients with FHH. Occasionally, in primary hyperparathyroidism, PTH concentration will be inappropriately within the normal range compared to the serum calcium concentration. In all other conditions, PTH will be suppressed by hypercalcemia.
3. If no obvious malignancy is present and PTH concentration is not increased, the possibility of vitamin D intoxication or granulomatous disease should be evaluated further with an analysis of calcidiol and calcitriol concentration. An increased calcidiol concentration is seen with the ingestion of either vitamin D or calcidiol. An elevated calcitriol concentration is observed with calcitriol ingestion, granulomatous disease, lymphoma, and primary hyperparathyroidism.
4. As a final step, if calcitriol concentration is increased without an apparent cause, occult granulomatous disease can be evaluated with a hydrocortisone suppression test. After administration of 40 mg of hydrocortisone every 8 hours for 10 days, the hypercalcemia will resolve if it is the result of granulomatous disease.
D. Treatment of hypercalcemia varies depending on the severity of the serum calcium elevation. It is directed at increasing urinary calcium excretion, inhibiting bone resorption, and decreasing intestinal calcium absorption.
1. Urinary calcium excretion is increased by first expanding the ECF volume and, subsequently, administering loop diuretics. Calcium reabsorption in the proximal tubule is passive and parallels sodium reabsorption. ECF volume contraction, therefore, increases proximal sodium reabsorption and helps maintain hypercalcemia. Patients with hypercalcemia are often volume contracted. Hypercalcemia decreases sodium reabsorption in the thick ascending limb of the loop of Henle through activation of the calcium-sensing receptor, and it also antagonizes the effects of antidiuretic hormone. In the setting of a reduced GFR, higher doses of loop diuretics may be required. In the presence of little or no renal function and severe hypercalcemia, hemodialysis is indicated.
2. An agent that inhibits bone resorption is often required when hypercalcemia is moderate or severe. In the acute setting, calcitonin is often helpful because of its rapid onset of action (2 to 4 hours). Calcitonin inhibits osteoclastic bone reabsorption and increases renal calcium excretion. It reduces serum calcium concentration, however, by only 1 to 2 mg/dL, and tachyphylaxis often develops with repeated use. For these reasons, calcitonin should not be used as the sole agent to inhibit bone resorption.
a. Bisphosphonates are the agents of choice for the management of hypercalcemia due to bone resorption. These analogs of inorganic pyrophosphate are selectively concentrated in bone, where they interfere with osteoclast attachment and function. Bisphosphonates have a slow onset (2 to 3 days) and long duration of action (several weeks). Caution should be exercised in patients with milk-alkali syndrome. These patients do not have a defect in bone turnover and are susceptible to hypocalcemia with treatment, and post treatment hypocalcemia may be exacerbated by bisphosphonates.
Pamidronate is given at a dose of either 60 or 90 mg intravenously over 4 hours. If serum calcium concentration is ≤13.5 mg/dL, 60 mg is given. If serum calcium concentration is greater than 13.5 mg/dL, 90 mg is administered. Serum calcium concentration gradually falls over the ensuing 2 to 4 days. A single dose is usually effective for 1 to 2 weeks. In most patients, serum calcium concentration normalizes after 7 days.
Zolendronic acid is now the most commonly used bisphosphonate because it can be given intravenously, which avoids esophageal damage from oral doses, and can be administered over a short interval (4 mg over 15 minutes). It is administered every 3 to 4 weeks if needed and it may be longer lasting than pamidronate. The dose must be adjusted in patients with renal dysfunction as follows given the creatinine clearance: greater than 60 mL/minute—4 mg; 50 to 60 mL/minute—3.5 mg; 40 to 49 mL/minute—3.3 mg; 30 to 39 mL/minute—3.0 mg; less than 30 mL/minute—no data available. The manufacturer recommends that the drug be discontinued if the serum creatinine concentration increases ≥0.5 mg/dL above a normal baseline or greater than 1.0 mg/dL in those with a serum creatinine concentration ≥1.4 mg/dL.
Bisphosphonates are associated with significant toxicity including focal glomerular sclerosis and acute kidney injury. Most of these cases occurred in patients with preexisting CKD or when recommended doses were exceeded. Multiple myeloma patients are at particular risk because kidney disease is a common complication and management of osteoporosis and/or hypercalcemia may require higher than recommended doses. In addition, when bisphosphonates are used long term in patients with malignancy, especially multiple myeloma and breast cancer, they are associated with osteonecrosis of the jaw. Most of these patients have had recent tooth extraction or surgical tooth removal. Radiation to the jaw also increases the risk of osteonecrosis.
b. Gallium nitrate inhibits bone resorption by decreasing the acid secretion of osteoclasts and also enhancing hydroxyapatite
crystallization of bone. It is an additional agent that can be employed to treat hypercalcemia of malignancy. It is administered as a continuous infusion at a dose of 100 to 200 mg/m2 for 5 consecutive days. Gallium nitrate has a significant risk of nephrotoxicity and should not be administered to patients with serum creatinine concentrations above 2.5 mg/dL or when other nephrotoxic drugs are also used such as iodinated contrast, aminoglycosides, or cisplatinum. It is probably best reserved for patients who have not responded to more conventional agents. A summary of treatment options is shown in Table 5-2.
crystallization of bone. It is an additional agent that can be employed to treat hypercalcemia of malignancy. It is administered as a continuous infusion at a dose of 100 to 200 mg/m2 for 5 consecutive days. Gallium nitrate has a significant risk of nephrotoxicity and should not be administered to patients with serum creatinine concentrations above 2.5 mg/dL or when other nephrotoxic drugs are also used such as iodinated contrast, aminoglycosides, or cisplatinum. It is probably best reserved for patients who have not responded to more conventional agents. A summary of treatment options is shown in Table 5-2.
c. Cinacalcet is a calcimimetic agent with a unique mechanism of action. Though it has no structural similarity with elemental calcium it can bind to the receptor and cause allosteric activation. Calcium-sensing receptors are located in the gastrointestinal tract, kidney, bone, and parathyroid glands. Allosteric activation results in a reduction of PTH release, less calcium release from bone, less intestinal calcium absorption, and an increase in renal calcium excretion. This results in a lowering of calcium and PTH levels. The lower PTH level’s effect on serum phosphorus concentration is dependent on renal function. In dialysis patients with no renal function, decreased PTH levels can reduce phosphorus concentration by limiting bone resorption. In patients with significant renal function, such as renal transplant recipients with persistent tertiary hyperparathyroidism, the initial phosphorus concentration can be low because of persistently elevated PTH levels. Treatment with cinacalcet will reduce PTH levels and reduce renal phosphate wasting and increase rather than decrease serum phosphorus levels in these patients. The brain also has calcium-sensing receptors and a significant dose-limiting side effect is nausea. Cinacalcet is approved
for use in the United States for treatment of secondary and tertiary hyperparathyroidism and parathyroid carcinoma. In Europe, it is also approved for the medical treatment of primary hyperparathyroidism.
for use in the United States for treatment of secondary and tertiary hyperparathyroidism and parathyroid carcinoma. In Europe, it is also approved for the medical treatment of primary hyperparathyroidism.
Table 5-2. Treatment of Hypercalcemia |
|---|









