I. DEFINITION AND RECOGNITION OF ACUTE KIDNEY INJURY (AKI).
AKI, formerly known as acute renal failure, is a sudden decrease in kidney function characterized by a reduction in the glomerular filtration rate (GFR). AKI may occur in patients with previously normal renal function or patients with chronic kidney disease (CKD); in either case, the clinical approach to find and treat the cause remains similar. Criteria to diagnose AKI have been established by the Acute Kidney Injury Network (AKIN) and the
Risk,
Injury,
Failure,
Loss,
End-stage kidney disease (RIFLE) criteria (
Table 10-1). The AKIN and RIFLE classifications convey the concept that AKI is not only significant when it requires renal replacement therapy (RRT), but that it is a spectrum ranging from early disease to long-term failure. Based on the AKIN and RIFLE criteria, the definition of AKI is as follows: 1) an increase in serum creatinine from baseline by =0.3 mg/dL within 48 hours, or 2) an increase in serum creatinine =1.5 times baseline which is known or presumed to have occurred within the prior 7 days, or 3) urine volume <0.5 mL/kg/hour for 6 hours (as summarized in
Table 10-2). For example, an increase in serum creatinine from 2.0 to 2.3 mg/dL within 48 hours is diagnostic of AKI; similarly, an increase from 1.0 to 1.3 within 48 hours is diagnostic of AKI. The AKIN and RIFLE criteria have been validated in multiple studies. Furthermore, an increase in serum creatinine by 0.3 mg/dL is associated with an independent increased risk of mortality. The recent Kidney Disease/Improving Global Outcomes (KDIGO) Clinical Practice Guidelines for AKI definition is in agreement with this definition (
Table 10-2).
A. Serum Creatinine as a Marker of AKI and GFR. Normal serum creatinine is 0.6 to 1.2 mg/dL and is the most commonly used parameter to assess kidney function. Unfortunately, the correlation between serum creatinine concentration and GFR may be confounded by several factors.
1. Creatinine Excretion is Dependent on Renal Factors Independent of Function. Certain medications such as trimethoprim or cimetidine interfere with proximal tubular creatinine secretion and may cause a rise in serum creatinine without a fall in GFR (
Table 10-3). Once filtered, creatinine cannot be reabsorbed.
2. Serum Creatinine is Dependent on Nonrenal Factors Independent of Kidney Function. For example, creatinine production is dependent on muscle mass. Muscle mass declines with age and illness. Therefore, a serum creatinine of 1.2 mg/dL in an elderly, 40-kg patient with cancer and wasted muscles may represent a severely impaired GFR, whereas a serum creatinine of 1.2 mg/dL in a 100-kg weightlifter with large muscle mass may represent a normal GFR. Serum creatinine is also
dependent on other factors such as nutritional status, infection, volume of distribution, age, gender, race, body habitus, presence of amputations, malnutrition, and diet.
3. Creatinine Production and Excretion Must Be in a Steady State Before Creatinine Levels Accurately Reflect the Decline in Kidney Function. The most commonly used formulae to estimate GFR, in a steady state, are the Cockcroft-Gault, Modification of Diet in Renal Disease (MDRD), the modified MDRD, and the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equations. In a steady state, the CKD-EPI equation is the most reliable estimate of kidney function. However, all of these formulae need to be used with caution in estimating kidney function in patients with AKI. For example, after an acute insult, it takes several days for creatinine excretion and production to reach a steady state and kidney function will be worse than what the formulae suggest. For example, if a 60 kg, 30-year-old woman with a serum creatinine of 1.0 mg/dL suddenly loses all renal function, her serum creatinine may only rise to 1.8 mg/dL after 1 day. By CKD-EPI, her GFR is 37 mL/minute; by Cockcroft-Gault it is 43 mL/minute, but it is actually 0 mL/minute. For reference, the formulae for Cockcroft-Gault, MDRD, modified MDRD, and CKD-EPI are listed below.
a. Cockcroft-Gault Formula
GFR = [([140 – age (years)] × lean body weight in kg)/(serum Cr × 72)] × (0.85 if female)
b. MDRD Formula
GFR, in mL/minute/1.73 m2 = 170 × (serum Cr-0.999)
× (age-0.176) × (BUN-0.170)
× (serum albumin+0.318)
× (0.762 if female)
× (1.180 if black)
where serum Cr (creatinine) and blood urea nitrogen (BUN) are in mg/dL; serum albumin is in g/dL.
c. Modified MDRD Formula
GFR, in mL/minute/1.73 m2 = 186.3 × (serum Cr–1.154)
× (age-0.203) × (0.742 if female)
× (1.21 if black)
d. CKD-EPI
GFR, in mL/minute = 141 × min(SerumCreat/kappa, 1)alpha × max(SerumCreat/kappa, 1)-1.209
× 0.993Age × Sex × Race
For females, the following values are used: Sex = 1.018; alpha = -0.329; kappa = 0.7. For males, the following values are used: Sex = 1; alpha = -0.411; kappa = 0.9.
e. Creatinine clearance (CrCl) may be measured in the acute setting to give an estimate of kidney function; more reliable results will be obtained when creatinine production and excretion are in a steady state. Steady state may be suggested when the creatinine reaches its peak and then stabilizes (e.g., if creatinine (mg/dL) is 1.0 at baseline, 2.0 on day 2, 4.0 on day 3, and 4.0 on the subsequent days, one may reasonably conclude that a steady state has been achieved at a creatinine of 4.0). Normal ranges for CrCl are 120 ± 25 mL/minute
for men and 95 ± 20 mL/minute for women. The formula for CrCl performed on a 24-hour urine collection is as follows:
CrCl = [urine creatinine (mg/dL) × urine volume (mL/24 hours)] /[serum Cr (mg/dL) × 1,440 minutes]
When the reduction in kidney function is severe, both CrCl and urea clearance may be determined on the same 24-hour urine collection; the average of CrCl and urea clearance may be a more accurate assessment of kidney function than CrCl alone (due to the increase in creatinine secretion that may occur with kidney dysfunction which will increase the amount of creatinine in the urine not related to GFR).
B. BUN as a Marker of AKI and GFR. Normal BUN is 8 to 18 mg/dL. An increase in BUN typically accompanies a rise in serum creatinine in the setting of AKI. Urea is filtered, but not secreted. Increased reabsorption of urea by the proximal tubule and arginine vasopressin (AVP)-sensitive urea transporters in the collecting duct occurs in states of volume depletion. In this setting, BUN can rise without a rise in creatinine, resulting in a BUN to serum creatinine ratio that is greater than 20.
BUN levels are affected by multiple factors not related to GFR. Because BUN production is related to protein metabolism, an increase in BUN without a decline in GFR may occur with hypercatabolic states, protein loading, upper gastrointestinal (GI) bleeding, and high-dose steroid administration. Conversely, a low BUN may be present in the setting of reduced GFR in patients who are on a low-protein diet, are severely malnourished, or have severe liver disease.
C. Cystatin C as a Marker of AKI and GFR. Cystatin C is a protein produced by all nucleated cells. It is freely filtered by the glomerulus, completely reabsorbed by the proximal tubules, and is not secreted by the renal tubules. Therefore, some of the limitations of serum creatinine, for example, effect of muscle mass, are not a problem with cystatin C. In AKI, changes in cystatin C occur sooner after changes in kidney function than serum creatinine. In studies, serum cystatin C correlated better with GFR than did serum creatinine and was diagnostically superior to creatinine especially in patients with liver cirrhosis. Cystatin C is best measured by an immunonephelometric assay, but is not yet routinely measured except in patients in whom serum creatinine is judged to be a poor marker of renal function, for example, liver cirrhosis, and in patients with reduced muscle mass.
D. Biomarkers of AKI. A biomarker that is released into the blood or urine by the injured kidney (analogous to troponin release by injured myocardial cells after myocardial ischemia) is a more sensitive and specific marker of AKI than BUN and serum creatinine. Urinary interleukin-18 (IL-18), neu trophil gelatinase-associated lipocalin (NGAL), kidney injury molecule-1 (Kim-1), and tubular enzymes have been found to increase 1 to 2 days before serum creatinine in patients with ischemic AKI. Higher levels of IL-18, NGAL, KIM-1, and liver fatty acid binding protein (L-FABP) also predict worsening AKI and death. Many studies are in progress to develop biomarkers of AKI that are superior to BUN and serum creatinine and will allow the early detection of AKI.
E. Distinguishing AKI from CKD. Distinguishing AKI from CKD may be challenging. Laboratory findings such as hyperphosphatemia, hypoalbuminemia, and hyperkalemia are unreliable factors to distinguish AKI from CKD and may be present in either case. Symptoms such as nausea, vomiting, and malaise may also occur in AKI or CKD. Potential methods to distinguish between the two include the following:
1. Old Records. The most reliable way to distinguish AKI from CKD is an evaluation of old records. Increased BUN or serum creatinine documented months earlier and/or a history of kidney disease suggests that the renal failure is chronic.
2. Renal Ultrasonography. As summarized in
Table 10-4, ultrasound may be a useful technique to distinguish AKI from CKD. Increased echogenicity (i.e., the kidney appears brighter than the normal liver) may occur in either AKI or CKD (echogenicity may also be normal in AKI or CKD); however, decreased kidney length or cortical thinning do not occur in AKI. Therefore, decreased kidney length and/or cortical thinning suggests that CKD is present. It is important to note that since AKI is common in patients with CKD, the presence of small kidneys or a thin cortex does not necessarily exclude the possibility that AKI is also present. For reference, “normal” kidney size is dependent on age. For example, at age 55, normal kidney length is approximately 11 cm; at age 75, normal kidney length is approximately 10 cm (although it is currently unknown whether the decrease in kidney length that is observed in aging is “normal” or represents undetected CKD). Normal cortex is approximately 1 cm.
3. Anemia. Normochromic normocytic anemia is common in patients with CKD and a GFR less than 30 mL/minute; in patients with a GFR of 30 to 44 mL/minute, only approximately 20% of patients have anemia. Therefore, with a GFR of 30 mL/minute or below, the absence of anemia suggests that the decline in renal function may be acute. In some etiologies of CKD (e.g., autosomal dominant polycystic kidney disease), however, anemia may be absent. In some etiologies of AKI, anemia may be present, for example, hemolytic uremic syndrome (HUS) or thrombotic thrombocytopenic purpura (TTP). Thus, the presence or absence of anemia must be interpreted in context with other clinical indicators when considering the diagnosis of AKI versus CKD.
F. Urine Output in AKI. AKI is typically described as either oliguric or nonoliguric. Oliguria is defined as a urine output of less than 400 mL/day; 400 mL is the minimum amount of urine that a person in a normal metabolic state must excrete to get rid of the daily solute production. For example, a person with a daily solute production of 500 mOsm who concentrates urine to a maximum of 1,200 mOsm/L would need to pass approximately 400 mL of urine per day to excrete the daily solute production (i.e., 500 mOsm/1,200 mOsm/L = 417 mL of urine per day).
Anuria is defined as a lack of urine obtained from a bladder catheter; it has a short list of potential causes. It is most often caused by complete bilateral urinary tract obstruction, urinary tract obstruction in a solitary kidney, and shock. Less common causes are HUS and rapidly progressive glomerulonephritis (RPGN), particularly anti-glomerular basement membrane (GBM) antibody disease; bilateral renal arterial or venous occlusion can also cause anuria.
II. CLASSIFICATIONS OF AKI: DEFINITIONS AND CAUSES.
AKI is classified as either intrinsic renal or postrenal. Prerenal azotemia may also cause a decline in GFR, which is reflected by increased serum creatinine and BUN.
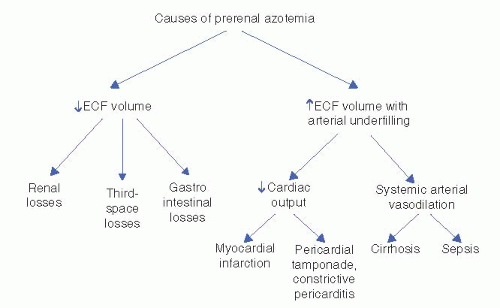
A. Prerenal Azotemia (
Fig. 10-1). Prerenal azotemia is a fall in the GFR due to reduced renal perfusion in which minimal cellular damage to the kidney has occurred. Urine sediment is typically bland and hyaline casts may be present. Essential to this diagnosis is that renal function returns to normal within
24 to 72 hours of correction of the hypoperfused state. Prerenal azotemia occurs in the following situations:
1. Total Intravascular Volume Depletion. This condition can occur in a number of settings where intravascular volume is reduced and may be secondary to
a. Hemorrhage
b. Renal fluid loss
Excessive diuresis (e.g., diuretics)
Osmotic diuresis (e.g., glucosuria, mannitol administration)
Primary adrenal insufficiency (i.e., hypoaldosteronism)
Salt-wasting nephritis
Diabetes insipidus
c. GI fluid loss
d. Skin fluid loss
Burns
Excessive sweating
Hyperthermia
e. Third-space fluid loss
2. Effective Volume Depletion from Arterial Underfilling. Arterial underfilling is a state in which intravascular volume is actually normal (or even increased) but circulatory factors are inadequate to maintain renal perfusion pressure. Underfilling may be due to either a decrease in cardiac output or arterial vasodilatation and may occur in a number of clinical settings:
a. Reduced cardiac output
Acute decompensated heart failure (ADHF) (previously referred to as congestive heart failure)
Cardiogenic shock (e.g., acute myocardial infarction)
Pericardial effusion with tamponade
Massive pulmonary embolism
b. Peripheral vasodilatation
3. Intrarenal Hemodynamic Changes
a. Glomerular afferent arteriole vasoconstriction (preglomerular effect)
b. Glomerular efferent arteriole vasodilatation (postglomerular effect)
B. Postrenal AKI. Postrenal AKI is caused by the acute obstruction of the flow of urine. Urinary obstruction of both ureters, the bladder, or the urethra may cause postrenal AKI. Patients most at risk for postrenal AKI are elderly men, in whom prostatic hypertrophy or prostatic cancer may lead to complete or partial obstruction of urine flow. In women, complete urinary tract obstruction is relatively uncommon in the absence of pelvic surgery, pelvic malignancy, or previous pelvic irradiation. The causes of postrenal AKI include the following:
1. Bilateral Ureteral Obstruction or Unilateral Obstruction in a Solitary Kidney (Upper Urinary Tract Obstruction)
a. Intraureteral
Stones
Blood clots
Pyogenic debris or sloughed papillae
Edema following retrograde pyelography
Transitional cell carcinoma
b. Extraureteral
c. Bladder neck/urethral obstruction (lower urinary tract obstruction)
Prostatic hypertrophy
Prostatic and bladder carcinoma
Autonomic neuropathy or anticholinergic agents causing urinary retention
Urethral stricture
Bladder stones
Fungal infection (e.g., fungus balls)
Blood clots
C. Intrarenal or Intrinsic AKI. In contrast to prerenal azotemia and postrenal AKI, the disorders listed here represent problems that originate within the kidney itself. These problems may be vascular, glomerular, interstitial, or tubular. The diseases may be primary renal or part of a systemic disease. The course of AKI in these situations cannot be changed by manipulating factors outside the kidney (e.g., performing volume repletion, improving cardiac function, correcting hypotension, or removing obstruction).
1. Vascular. Vascular disorders causing AKI are classified based on the size of the vessels involved.
a. Large- and medium-sized vessels
Renal artery thrombosis or embolism
Operative arterial cross-clamping
Bilateral renal vein thrombosis
Polyarteritis nodosa
b. Small vessels
2. Glomerular. Glomerular diseases are typically categorized based on urine findings as either nephrotic or nephritic.
a. Nephrotic glomerular disorders are characterized by large proteinuria (greater than 3 g in 24 hours) and minimal hematuria. Nephrotic glomerular disorders are uncommonly associated with AKI, but may occur in minimal-change disease or focal segmental glomerulosclero sis (FSGS), particularly collapsing FSGS.
b. Nephritic glomerular disorders (glomerulonephritis) are character ized by hematuria and proteinuria (typically 1 to 2 g in 24 hours). Patients with known glomerulonephritis may develop AKI; alterna tively, glomerulonephritis may present as AKI. Rapidly progressing Glomerulo Nephritis (RPGN), also called crescentic nephritis, should be suspected in a patient with a rising creatinine, hematuria, and pro teinuria. RPGN is caused by injury to the glomerular capillary wall, which results in subsequent inflammation, fibrosis, and crescent for mation. Urgency is required to make the diagnosis of RPGN, because crescent formation can rapidly destroy the glomeruli; response to therapy is directly correlated with the percentage of glomeruli having crescents. If RPGN is suspected, a biopsy should be performed as soon as possible as waiting even a few days can result in irreversible loss of kidney function. Because the diagnosis is typically made by renal biopsy, the causes of glomerulonephritis and RPGN are classi fied according to immunofluorescence staining on renal biopsy.
i. Diseases with Linear (anti-GBM) Immune Complex Deposition
ii. Diseases with Granular Immune Complex Deposition
Acute postinfectious glomerulonephritis
Lupus nephritis
Infective endocarditis
Immunoglobulin (Ig) A glomerulonephritis
Henoch-Schönlein purpura
Membranoproliferative glomerulonephritis
Cryoglobulinemia
iii. Diseases with No Immune Deposits (Pauci-immune)
Granulomatosis with polyangiitis (GPA), (formerly known as Wegener’s granulomatosis)
Microscopic polyangiitis (MPA)
Churg-Strauss syndrome (CSS)
Idiopathic crescentic glomerulonephritis
3. Interstitium. AKI from an interstitial cause is known as acute interstitial nephritis (AIN). The primary histologic lesion of AIN is marked edema of the interstitial space with a focal or diffuse infiltration of the renal interstitium with inflammatory cells (lymphocytes and/or eosinophils). AIN (also called acute tubulointerstitial nephritis) is most commonly due to drug hypersensitivity, but may also be a consequence of infections or systemic disease (e.g., systemic lupus erythematosus).
a. Drug-Induced AIN. More than 100 drugs have been implicated in drug-induced AIN. Some of the drugs most commonly associated with AIN are as follows:
Antibiotics (e.g., methicillin, cephalosporins, rifampicin, sulfonamides, erythromycin, and ciprofloxacin)
Diuretics (e.g., furosemide, thiazides, chlorthalidone)
NSAIDs
Anticonvulsant drugs (e.g., phenytoin, carbamazepine)
Allopurinol
b. Infection-Associated AIN
Bacterial (e.g., Staphylococcus, Streptococcus)
Viral (e.g., cytomegalovirus, Epstein-Barr virus)
Tuberculosis
4. Tubular. Acute tubular necrosis (ATN) is characterized by an abrupt decrease in GFR due to proximal tubular dysfunction most commonly caused by ischemic AKI or nephrotoxic AKI. Although this type of renal injury has long been designated ATN, the term is a misnomer because, in many cases, true necrosis of tubular cells is not present on histologic examination. Most of the renal biopsies are, however, late and therefore could miss early tubular necrosis. The tubules may demonstrate morphologic changes of sublethal injury (e.g., swelling, vacuolization, loss of brush border, apical blebbing, and loss of basolateral infoldings). Loss of viable and nonviable tubular epithelial cells into the urine also occurs. The continued presence of renal blood flow and reversibility of tubular dysfunction is compatible with the recov ery of renal function that is seen in some patients with ischemic or nephrotoxic AKI.
Ischemic AKI is a consequence of reduced blood flow to the kidneys, which results from a decreased total blood volume or arterial underfilling with a redistribution of blood away from the kidney. Ischemic AKI is seen most commonly after septic or hemorrhagic shock. Nephrotoxic AKI is most commonly caused by aminoglycoside antibiotics and radiocontrast dye. In most cases, the insults are multifactorial.
Causes of ischemic or nephrotoxic AKI include the following:
a. Renal Ischemia
b. Nephrotoxic Drugs
Aminoglycoside antibiotics
Amphotericin B
Pentamidine
Foscarnet
Acyclovir
Indinavir
Antineoplastic agents (e.g., cisplatin)
Radiocontrast dye
Organic solvents (e.g., carbon tetrachloride)
Ethylene glycol (antifreeze)
Anesthetics (enflurane)
Oral sodium phosphosoda used for bowel preparation for colonoscopy can cause acute phosphate nephropathy resulting in acute nephrocalcinosis
c. Endogenous Toxins
Myoglobin (e.g., rhabdomyolysis)
Hemoglobin (e.g., incompatible blood transfusion, acute falciparum malaria)
Uric acid (e.g., acute uric acid nephropathy)
5. Sepsis. Sepsis is the most common cause of AKI in the intensive care unit (ICU). The pathophysiology of AKI in sepsis is complex, and many aspects of the cause of renal function decline in sepsis remain controversial. Although previously thought to be similar to ischemic AKI, it is now understood that septic AKI is a separate entity from ischemic AKI—although ischemic AKI may ultimately occur in severe sepsis or shock from reduced renal blood flow. Renal function decline in sepsis is likely due to a combination of vascular factors (affecting autoregulation and resulting in decreased GFR) as well as intrinsic tubular damage.
 The Edematous Patient: Cardiac Failure, Cirrhosis, and Nephrotic Syndrome
The Edematous Patient: Cardiac Failure, Cirrhosis, and Nephrotic Syndrome
 The Patient with Urinary Tract Infection
The Patient with Urinary Tract Infection
 The Patient with Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
The Patient with Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
 The Patient with Glomerular Disease or Vasculitis
The Patient with Glomerular Disease or Vasculitis
 The Patient with Kidney Disease and Hypertension in Pregnancy
The Patient with Kidney Disease and Hypertension in Pregnancy
 The Patient with Hypertension
The Patient with Hypertension



