Stomach and Proximal Duodenum: Inflammatory and Miscellaneous Disorders
Stomach and Proximal Duodenum: Inflammatory and Miscellaneous Disorders
CLASSIFICATION OF GASTRITIS AND GASTROPATHY
Though no classification of gastritis satisfies everyone, the overall goal of any classification is to help clear thinking and be clinically useful. Inevitably much of the early thinking regarding gastritis was centered on “peptic ulcer disease” (PUD). Ignorance regarding the role of both Helicobacter and medications gave rise to theories that were to some extent flawed, yet they still dominate traditional teaching. Gastritis was considered physiologic and intestinal metaplasia an aging phenomenon. We also need to recall that
1. Gastritis originally meant “redness”—which now is usually associated with a gastropathy rather than gastritis; conversely, most histologic gastritis has a normal endoscopic appearance.
2. Many disorders that are characterized by abnormal endoscopy also have a typical biopsy appearance. From a classification viewpoint are these best considered from an endoscopic or histologic viewpoint? Most classifications can only be viewed from one vantage point.
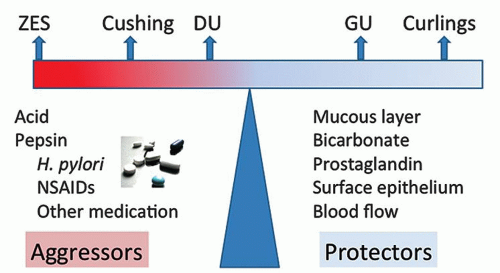
3. From a clinical viewpoint, “ulcers” have played a major role in gastric disease because of the symptoms with which they or their complications are associated (pain, bleeding, perforation, and obstruction/stenosis). However, the term “peptic ulcer disease” has been in common parlance for decades, with the implication that this is associated with acid, the “proof” being that symptoms are markedly ameliorated with therapy, whether antacids, H2-receptor antagonists, or proton pump inhibitors (PPIs). In the early 1980s, it was ultimately shown that some ulcer disease, especially in the duodenum, was related to Helicobacter pylori, so that its eradication virtually guaranteed that duodenal ulcer, the archetypal peptic ulcer, would not recur. Thus PUD changed from being primarily acid related to primarily bacterial, or a combination of both.
4. Nonsteroidal anti-inflammatory drugs (NSAIDs), aspirin (acetylsalicylic acid—ASA), and other medications now play a huge role in gastric pathology. While the introduction of NSAIDs around 1970 was a major step forward therapeutically, it came at a price that included numerous gastrointestinal (GI) side effects. Prior to this time, ASA had been “the” analgesic and antipyretic of choice. Bayer introduced ASA in the market around 1900, and within a decade or two this “wonderdrug” was present in virtually every household in the more developed countries, and used widely for numerous ailments—colds, coughs, headaches, migraines, and all arthritides. Yet the erosive, ulcerative, and bleeding diathesis associated with this drug was not widely appreciated. In retrospect, from about 1900 on, many “peptic ulcers” may well have been as much ASA associated as Helicobacter associated, and this association even creeps, almost inadvertently, into case reports back in the 1950s.1 So while we typically think of “peptic ulcer disease” historically as unrecognized Helicobacter infection, ASA was very likely a major contributor. This continued until acetaminophen/paracetamol/Tylenol came into the market in to the 1960s. Further, it is now well recognized that, especially in the very young2, 3 and elderly,4 not only that NSAIDs are likely “the” culprit irrespective of the presence of H. pylori, but that the risk of complications such as bleeding (and therefore the erosions and ulcers that bleed) can be largely prevented using PPIs. Thus, historically, the disease we consider to be “peptic ulcer disease” may have been as much NSAID/ASA associated as Helicobacter associated, especially in the presence of abundant acid.
5. Historically, alcohol, which not only has a social role in many societies but is also an analgesic in large doses, has been around much longer than any other gastric damaging agent except for Helicobacter, and produces histologic changes similar to NSAIDs (i.e., a chemical/reactive gastropathy). From around 1900 on when aspirin became available, the big three, became Helicobacter, alcohol, and ASA, and from 1970 on, NSAIDs was added to these.
6. The notion of “peptic ulcer disease” and “no acid— no ulcer” is therefore likely true in that in the major causes of gastroduodenal erosions and ulcers, namely, Helicobacter and NSAIDs/ASA, and other medications or chemicals, especially alcohol, the presence of acid facilitated the development of injury caused by these agents. Although the nature of the interaction of these common causes of peptic ulcer is still unclear, it would make most sense if, when antral-predominant H. pylori is present, that the risk of NSAID/ASA and alcohol-induced damage was increased, but that when the organism spread proximally, resulting in a decrease in acid output, that there may well be a degree of protection from NSAID/ASA, and possibly alcohol-associated damage (Table 13-1).
Current Classification of Gastritis
Until the early 1970s, chronic gastritis was classified into three main varieties (superficial, atrophic, and hypertrophic) as suggested by Schindler in 19395 (Table 13-2). Wood as well as Schindler later concluded that chronic hypertrophic gastritis is a variation of normal mucosal function.6, 7 Thus, chronic gastritis was classified as superficial or atrophic.
Whitehead’s classification was the first to understand the importance of noting location, and grading the depth and degree of inflammation and the presence or absence of both intestinal and pseudopyloric metaplasia, separating them from atrophic changes8 (Table 13-2). This really formed the basis of all subsequent morphologic classifications of gastritis. In 1973, Strickland and Mackay classified gastritis based on detecting parietal cell (PC) antibodies, clarifying the etiology of autoimmune gastritis (AIG) (type A) despite the fact that these can develop in Helicobacter infected patients. It is associated with atrophic changes in body and fundic (oxyntic) mucosa. Antral predominant gastritis was type B. Glass and Pitchumon added type AB into Strickland-Mackay classification to encompass cases that did not fit type A or type B, essentially pangastritis.
Table 13-1 ABC Classification of Gastritis
Autoimmune
Pernicious anemia
Bacterial
Bugs including post-Rx effects:
Helicobacter pylori
Enterococcus
Syphilis
Chemical
Bile reflux
Drug-associated/Iatrogenic
NSAIDs/ASA
Anti-platelet medications
Chemotherapy/GVHD
Iron
Alcohol
Eosinophilic
Eosinophilic gastritis/gastroenteritis
Food allergies, medications
Focal
Crohn’s disease
Granulomatous
Tuberculosis
Sarcoid
Crohn’s disease
Foreign body
Helicobacter pylori
Hypertrophic (big folds)
“Ménétrier’s disease”
Lymphocytic gastritis
Eosinophilic gastritis
Gastric varices
Gastritis cystica profunda
Lymphoma (MALT)
Gastric adenocarcinoma
Helicobacter pylori gastritis (lymphocytic), CMV
Zollinger-Ellison syndrome
Multiple polyps/polyposis
Idiopathic
Juvenile (pediatric)
Follicular with H. pylori, CMV
Lymphocytic
Helicobacter pylori, celiac disease
Chronic erosive (varioliform) gastritis
Multifocal intestinal metaplasia with/without atrophic front
In atrophic gastritis, isolated
Modified from Wyatt JI, Dixon MF. Chronic gastritis—a pathogenetic approach. J Pathol. 1988;154(2):113-124.
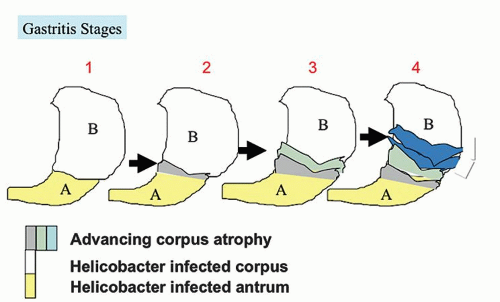
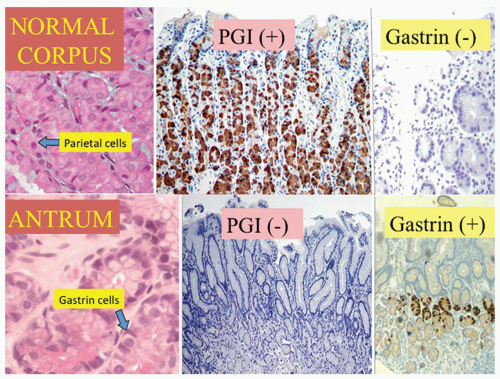
With the rediscovery of H. pylori (originally Campylobacter pylori) by Warren and Marshall in the early 1980s,9 it became clear that H. pylori is a principal component of most gastritides. In 1988, two classification systems emerged. That by Wyatt and Dixon incorporated reactive gastropathy (then called chemical gastritis/gastropathy) as the “C” of the ABC classification system, A being autoimmune and B being bacterial (=Helicobacter but, at that time, C. pylori).10 The same year, Correa proposed classifying gastritis based on clinical and etiopathogenetic information. He classified chronic gastritis into superficial gastritis, diffuse antral gastritis (DAG), usually Helicobacter associated and related to duodenal ulcer disease, diffuse corporal atrophic gastritis (autoimmune), and multifocal atrophic gastritis (MAG—considered to be “environmental”). MAG was related to intestinal-type adenocarcinoma and gastric ulcer, and intestinal metaplasia in the antrum and body.11 Diffuse corporal atrophic gastritis was often related to AIG and pernicious anemia, with inflammation and atrophy in the corpus and relative sparing of the antrum11, 12 (Fig. 13-1).
The Sydney system is the basis of most contemporary classifications of gastritis. Proposed by a group of European pathologists and clinicians (World Congress of Gastroenterology, Sydney, August 1990),12 it recommended incorporating the topography of gastric mucosal changes with the immunology and microbiology of the disease. The classification depends on separate assessment of the antrum and corpus by taking a minimum of two biopsies from the anterior and posterior walls of the respective gastric compartments as well as any specific lesions identified. An important feature is a standard three-tier grade of mild, moderate, and severe applicable to a selected number of morphologic variables. As a broad guideline, each successive grade represents an increment in severity of about one-third. Graded variables included inflammation (acute and chronic), atrophy, metaplasia, and density of H. pylori. The Sydney system also expanded previous classifications by adding a variety of other “special forms” of gastritides (collagenous, eosinophilic, granulomatous, lymphocytic, etc.).
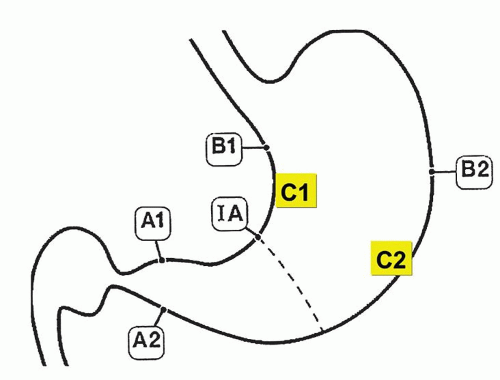
The Sydney classification was updated in 1994,13 which expanded the section on specific entities (special forms) and includes a 4-point visual analog (equivalent to none, mild, moderate, and severe) to aid with morphologic grading of inflammation and atrophy.13 Gastric atrophy is loss of normal glands, often with replacement by an epithelium that could be either native or metaplastic (Table 13-3). The score is an average from each region’s biopsies. Antral atrophy was the average score for atrophy from all antral biopsies and corpus atrophy (the average score for atrophy from all corpus biopsies).13 The updated Sydney classification depends on the separate assessment of the antrum and corpus. It needs a minimum of two biopsies from the lesser and greater curvature of the respective gastric compartments as well as the incisura and any specific lesions identified (Fig. 13-2).13 On all occasions accurate grading depends on correctly oriented full-thickness mucosal biopsies. In practice, other than for academic studies, grading is rarely required.
Figure 13-1. Prototypes of gastritis pattern predict disease outcome. In practice, all tend to have some degree of both antral and corpus inflammation. Top left: Duodenal ulcer (DU) patients have antral predominant inflammation with little corpus inflammation. Bottom left: Pangastritis is seen in gastric ulcer (GU) patients. Corpus mucosa is inflamed and often extends into the specialized mucosa but still tends to be antral predominant. Top right: Pangastritis with atrophy is seen in patients with the intestinal type of gastric adenocarcinoma (CA). Bottom right: Corpus-predominant gastritis is usually seen in AIG or end-stage Helicobacter infection.
GASTRITIS
Gastritis (in its broadest sense) and its complications account for millions of doctors’ office visits each year. Symptoms are often associated with acute changes or complications described as mild upper abdominal discomfort, indigestion, heartburn, coated tongue, foul breath, and bad taste to more ominous symptoms such as loss of appetite, nausea, vomiting blood or coffee-ground material, diarrhea, and dark stools. Most patients with chronic gastritis have no symptoms. Even so, these symptoms are not specific and include broad differentials such as H. pylori infection, other infections, bile reflux, inflammatory bowel disease (IBD), and side effects of medications (Table 13-4). As treatment depends on the cause, it is important to know the cause for appropriate management. Occasionally, it may be necessary to list possible etiologies for gastric inflammation, rather that reporting “nonspecific chronic inflammation”—which is an unnecessarily complex term as all inflammation is “nonspecific,” so these words can always be omitted from reports without deleterious effect. If it is specific, the cause (e.g., Helicobacter) should be stated.
Figure 13-2. The updated Sydney biopsy protocol requires a minimum of two biopsies from the lesser and greater curvature of the respective gastric compartments as well as the incisura and any specific lesions identified. This identifies all of the patterns of gastritis illustrated in Figure 13-1, as well as estimating the extent of atrophy present, which often starts at the incisura/angulus (IA), affects the antrum (A1, A2), and then extends proximally to the oxyntic zone (B1, B2), so that, as antral inflammation extends proximally, biopsy site B1 is first affected, and B2 is the last site affected.
Distinctive (Specific) Types of Gastropathies
Gastropathies are biopsies in which epithelial (noninflammatory) changes predominate. The mucosa is often mucin depleted, causing it to appear red endoscopically (invariably interpreted by endoscopists as “gastritis” rather than areas of redness). They include biopsies with primary epithelial reactive changes (such as chemical/reflux (bile) gastropathy, chemotherapy effect) and a smaller subset of biopsies with predominant vascular pathology (such as gastric antral vascular ectasia [GAVE], portal hypertension gastropathy, Dieulafoy, and hemorrhagic/shock) (Table 13-4). Graft versus host disease (GVHD) is usually normal endoscopically.
Table 13-4 Classification by Predominant Histologic Change
Distinctive macroscopic (endoscopic) appearance with appropriate histology
Erosive and hemorrhagic
Varioliform gastritis
Watermelon stomach (GAVE)
Portal gastropathy
Hemorrhagic gastritis/gastropathy
Nonerosive
Nodular gastritis, children
Atrophic front, adults
Distinctive hypertrophic gastropathy
Reactive (Predominant Epithelial) Changes
Reactive (chemical/reflux-associated) gastropathy is a reaction to noninfectious irritants. This can be due to protracted exposure to bile and pancreatic juice (especially postgastric surgery14). The most infamous of irritants are NSAIDs, which include over-the-counter drugs such as aspirin and ibuprofen, and many prescription medicines. Other medications— such as bisphosphonates used for osteopenia, iron pills and irritants in food such as capsaicin in peppers and chilies and alcohol—can all cause this lesion.15 These irritants usually cause no clinical problems when taken for the short term, although endoscopic damage can be seen even with short-term use. However, regular (or excessive) use can lead to a more severe gastropathy as well as erosions and ulcers. With the increasing use of aspirin and other NSAIDs, and decreasing prevalence of Helicobacter, chemical/reactive gastropathy is increasingly seen in gastric biopsies, and may co-exist. Anti-platelet mediations also cause similar injury.
Pathogenesis: Aspirin is the best-studied NSAID, the mechanism of injury is inhibition of prostaglandin synthesis by inhibiting cyclooxygenase (COX) 1 and 2.16 Aspirin also changes the ability of the mucosa to maintain a pH gradient causing gastric acid back-diffusion with resultant mucosal injury.16 Further, its anticoagulant properties increase the risk of bleeding once erosions or ulcers are present. Conversely, some other NSAIDs have antiplatelet properties but do not possess this therapeutic anticoagulant effect. NSAIDs produce mucosal injury by both local and systemic effects.16 Newer NSAIDs are predominantly COX-2 inhibitors, which make them less likely to cause gastric injury and the risk of gastric (or duodenal) injury is reduced, but not abolished. A variety of antiplatelet medications are increasingly being implicated causing similar injury.
Histology: The histology of reactive gastropathies has both an acute and a chronic phase, although in practice it is often reported without qualifying it as acute or chronic. In some patients both are present together.
Reactive gastropathy. The morphologic changes that accompany ingestion of medications such as NSAIDs have been known for decades, 15 but they are now more commonly recognized.
In the acute phase, as in any reparative process, the main changes are
1. Mucin depletion—the amount of supranuclear mucin is markedly reduced or absent, so that at low power the cells appear more basophilic—often the most apparent low-power indication that this change is present
2. A reduction of the normal cell size so that the cells are frequently low columnar to cuboidal
3. A corresponding increase in nuclear size, and also an increase in hyperchromatism; nuclei that are normally compressed at the cell base markedly increase in size, and in conjunction with the smaller cell size cause a marked increase in nuclear-cytoplasmic ratio.
4. Because this appears to be a reparative, partly restitutional process, the number of cells and nuclei appears reduced; this results in nuclei being distinct and separated, one of the best indicators that this is not dysplasia. However, especially following erosions, the reactive changes can be more marked so that the nuclei become more open and vesicular with distinct nucleoli, and there may also be concomitant increase in nuclear hyperchromatism (Fig. 13-3).
5. Changes are usually most marked in the mucous neck region, and tend to decrease superficially. Interestingly, some of these changes appear on the surface, especially the mucin depletion, but most of the other changes are maximal in the mucous neck region suggesting that these are all a reaction to injury and not dysplasia. These changes can be easily missed in the fundic glands as the foveolae are shorter and changes can be mistaken for biopsy artifacts. Occasionally, the changes may extend deeper to involve the entire length of the oxyntic or antral glands (Fig. 13-3E).
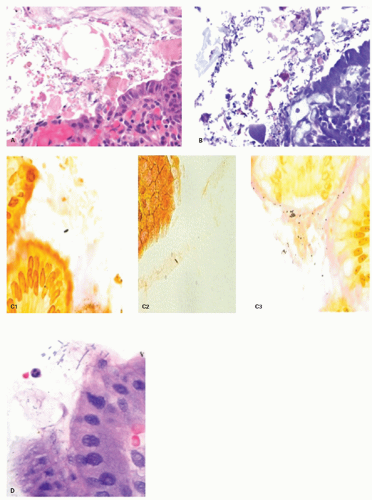
6. Erosions or ulcers may be present. When this occurs, careful examination of the erosion or ulcer base should be carried out for the presence of crystals or foreign material representing medications. Iron encrustation can readily be confirmed using Perl’s stain (Fig. 13-3). When erosions or ulcers occur, the immediately adjacent epithelium may be restituting, and appears attenuated as seen in any restitutional processes.
Figure 13-3. Reactive changes in gastric mucosa. A: Pit in which there is total mucin loss but nuclei are separated from each other. This is most marked superficially where the epithelial cells are more cuboidal and attenuated. Hints of mucin secretion are reappearing superficially (arrow) at the apex of the cell—an indication of maturation. Note the lack of any inflammation in the lamina propria in this biopsy. B: Similar features but there is more attenuation of epithelium superficially, and in the generative zone at the bottom nuclei are becoming stratified. The hyperchromatism associated with most dysplasias is absent. A modest chronic inflammatory infiltrate is present in the lamina propria but this disappears superficially. C: Chemical (NSAID) erosion. The attenuated epithelium is visible superficially with diffuse mucin depletion. Foveolar hyperplasia (corkscrewed pits) are visible, as is the normal architecture. At the surface the hyalinized zone is typical of NSAID damage. The lamina propria is largely empty indicating that this cannot be a Helicobacter-associated erosion.
Figure 13-3.(Continued)D: D1. An erosion with almost a pseudomembranous appearance. D2. The adjacent mucosa has typical reactive changes and scattered eosinophils predominate. E1: Further NSAID erosion with the superficial hyalinized band that approaches the muscularis mucosae and E2: Very reactive nuclei, again most marked at the bases of the pits, nuclei remain separated but here have a prominent nucleolus. More superficially nuclei are even more widely separated indicating restitution. Note also that these nuclear changes do not correspond to intestinal, foveolar, or pyloric dysplasia.
7. Occasionally there is focal edema in the lamina propria, which may also be devoid of inflammatory cells or have a predominantly acute or eosinophilic (sometimes both) infiltrate. A sparse chronic inflammatory infiltrate can also be seen, but in most biopsies chronic inflammation is usually conspicuous by its absence or minimal presence (Fig. 13-3), indicating that Helicobacter infection is not the etiology of the changes present.
In the chronic phase, other changes become apparent. Sometimes the acute and chronic phases coexist, sometimes only the chronic changes persist, and it is presumed that they followed the acute changes (Fig. 13-4).
1. Foveolar hyperplasia can develop that results in tortuosity of pits in the mucous neck region.
2. Proliferation of smooth muscle in the lamina propria above that normally seen
3. A degree of vasodilatation of capillaries with congestion and edema17, 18
4. If erosions have occurred, a degree of lamina propria fibrosis ensues.
Iron toxicity, especially in children, may result in gastric mucosal necrosis, sometimes with extension into the submucosa.19 The encrustation is often visible (Fig. 13-5).
Biopsies show reactive mucosal changes (mucin depletion, foveolar hyperplasia, and smooth muscle hyperplasia) without the severe inflammatory component seen in infectious gastritis (Fig. 13-5).
Figure 13-4. Chronic reactive gastropathy. The pits have foveolar hyperplasia, being elongated and have a corkscrew configuration (yellow arrows). There is hyperplasia of the smooth muscle fibers (blue arrow) that are normally found in the stomach. Note the lack of inflammation.
Foveolar hyperplasia appears to be a result of excessive cell exfoliation from the surface epithelium over a period of time and, accordingly, is likely to be seen in all types of active gastritis.17 Further, if the insult is ongoing, superimposed changes of acute reactive gastropathy may also be present, and this may include erosions. These histopathologic changes are not seen in all patients and when present are usually patchy (postgastrectomy states usually being more diffuse and therefore the exception). Pathology is more likely to be seen in biopsies obtained from incisura angularis.20 If biopsies are taken, then those from areas of endoscopic abnormality are preferred.
Gastric glands may be distorted and dilated, with an absence or paucity of plasma cells. In some areas there is no gland distortion, just simple thinning of the mucosa.21 Other features include stomal erosions,22 lipid islands, and intramucosal cysts21 (Fig. 13-6). Sometimes the cysts become large enough to be visible grossly and extend into the submucosa. These cysts have been labeled with a variety of names, including gastritis cystica polyposa and gastritis cystica profunda.23 Adenocarcinoma in the postoperative stomach has been reported in association with these cysts, but this association appears to be coincidental, especially because the cysts are so commonly found microscopically in the postoperative stomach.21, 24
Figure 13-5. Reactive gastropathy. Iron medication may result in gastric mucosal necrosis (H&E stain) with iron encrustation (Perl’s stain—right)
Toxic gastropathy. Changes can be seen characterized by vacuolated cells in the specialized mucosa. The cause is not always apparent but may be prominent in uremic patients, the vacuolation tending to occur in chief cells rather than PCs (Fig. 13-7).
Reactive Changes with Erosions in Helicobacter—One or Two Diseases?
It should be appreciated that while “reactive gastropathy” is usually applied to changes with minimal chronic inflammation, identical epithelial and lamina propria changes can be seen in other etiologies such as Helicobacter infections, especially if acute inflammation is present. However, in the presence of Helicobacter gastritis with relatively little acute inflammation, erosions are almost certainly not related to the underlying infection, and the possibility that the patient has medication-related erosions or ulcers superimposed on Helicobacter gastritis should be considered as the distinction can be made in many instances.25Helicobacter-type associated erosions invariably occur on a background of severe chronic active gastritis, so if this is not present they should always be viewed with suspicion, and a second etiology considered. Further, the nature of the erosion (see Fig. 13-3) can distinguish the two on biopsy, with a dense hyalinized band in the superficial mucosa being indicative of NSAID type-associated injury.26 Indeed, in patients with both diseases it is likely that a medication caused the damage.25, 26
Caveat: Severe (disproportionate) reactive changes resembling those seen in, for example, NSAID gastropathy in patients with only a modest chronic Helicobacter gastritis may well be related to medications rather than the concurrent Helicobacter infection. This is discussed subsequently.
Distinction of Reactive Changes from Dysplasia
It is imperative to distinguish reactive changes from dysplasia as they resolve when the acute insult is withdrawn. The most helpful feature is that at the surface there is invariably maturation in the form of small mucin droplets at the surface. While “bottom-up” dysplasia (dysplasia maximal in the pit bases) does occur, it is quite rare, so the diagnosis of dysplasia should only be made if the diagnosis is absolutely clear, and ideally conforms to one of the usual forms of foveolar dysplasia (see following chapter). The adage that dysplasia should never be diagnosed in the presence of overlying or adjacent ulcers, erosions, or restituting epithelium unless absolutely clear is a good one. Making a diagnosis of dysplasia under these circumstances is fraught with danger. Unless there is absolutely no diagnostic uncertainty, it is usually best to rebiopsy the area following antisecretory therapy (e.g., PPIs) to ensure that the changes persist when the erosions have healed. Fortunately, even if dysplasia is diagnosed and graded, most can be visualized and treated endoscopically (Fig. 13-8).
Figure 13-6. The postoperative stomach. A: Endoscopic view of a Billroth II stoma, which is typically red. Bile-stained fluid is refluxing into the gastric remnant. B: Biopsy specimen from the stoma of a Billroth II anastomosis. There is marked foveolar hyperplasia (corkscrew pattern), with minimal or no increase in the number of inflammatory cells. The epithelium in the surface and pits is dark and mucin depleted. Large intramucosal cysts are present. C: Fundic gland mucosa from the gastric body after Billroth II anastomosis. There is mild interfoveolar edema and marked foveolar hyperplasia with the corkscrew pattern, but an intact gland zone without increased numbers of inflammatory cells. D: Biopsy specimen from the greater curvature of the midbody region after Billroth II anastomosis. Many biopsy specimens in such patients simply show a thin fundic gland mucosa with a shallow epithelial gland zone, especially when the antrum has been removed as the gastrin drive for growth is lost. This specimen also shows subepithelial hemorrhage and edema in the interface between the pits and glands. It is not possible to exclude endoscope trauma as the cause of this finding.
Reactive gastropathy may be confused with dysplasia and may be one reason why some have reported large numbers of cases of dysplasia in the postoperative stomach.21 We suspect that the vast majority of these changes represent “regenerative atypia” rather than dysplasia. Highly reactive cytologic changes are seen in other conditions, such as in the mucosa adjacent to alcohol- and NSAID-induced erosions27 in some patients without erosions on NSAIDs17 and in the mucosa at or near-healed gastric ulcer sites. Though, at times, it can be challenging, atypical reparative changes can be distinguished from dysplasia (intraepithelial neoplasia or dysplasia) as discussed in the previous section.
Figure 13-7. Vacuolated cells that are prominent in toxic states, in this patient the association was uremia.
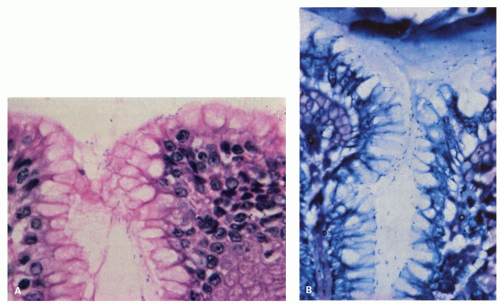
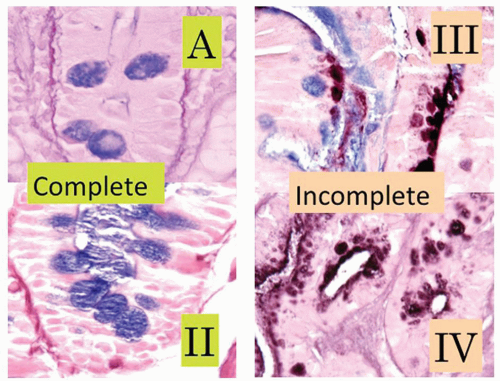
Reactive changes in intestinal metaplasia. It should also be recognized that gastric intestinal metaplasia, whether incomplete (residual foveolar epithelium admixed with goblet cells) or complete (goblet and absorptive cells with or without Paneth cells), can be subject to surface injury and reactive changes. However, the same principles apply regarding using surface maturation as an indicator of reactive changes and not diagnosing it in the presence of ulcers, erosions, or restituting epithelium. These can also be recognized by the presence of metaplasia in the adjacent mucosa. However, complete intestinal metaplasia starts with intestinal nuclei that are already considerably larger than native gastric mucosa. Nuclei in incomplete intestinal metaplasia are more open and vesicular with distinct small nucleoli. Reactive changes enhance all of these features, so this needs to be taken into account. All forms of reactive mucosa have both mucin depletion and enlarged pleomorphic nuclei occupying most of the cell, and may be accompanied by erosions or ulcer. The tip-off is the presence of (a) restituting mucosa (low cuboidal or columnar) with nuclei that are usually more widely separated than in normal mucosa, especially superficially, and (b) usually a degree of maturation superficially, to the degree that a diagnosis of dysplasia should be made very cautiously in the presence of active restitution. It is worthwhile to remember that in the bases of these pits, nuclei can overlap and be stratified and hyperchromatic causing confusion with adenoma/dysplasia.
Figure 13-8. A: Reactive changes versus dysplasia. Typical reactive changes with mucin depletion, but widely spaced nuclei and superficially attenuated epithelium (red arrows). These contrast with the closely packed stratified nuclei in the dysplastic crypts (blue arrows). B: Detail of (A). Reactive changes (red arrows) versus low-grade dysplasia (blue arrows).
Reporting reactive gastropathy: Minor degrees of superficial mucin depletion are relatively common, and it is unclear how much surface mucin depletion is required to report the changes, or indeed whether they can be seen physiologically. As a guide we do not report reactive changes unless the mucin droplet in the superficial epithelial cells (usually about 75%-80% of the cell) is <50%, but there are no data to support this. However, when reported we usually indicate the most common causes.
Reporting chronic changes: Usually mild chronic reactive changes alone, such as isolated foveolar hyperplasia, are not reported unless marked, as they tend to refer to events that happened at some point in the past, and it is unclear how long these changes take to reverse. If accompanied by acute changes of damage, then “reactive changes” covers both acute and chronic changes without the need to specify.
Clinical Implications: Of the millions of patients who every day ingest NSAIDs/ASA, only about 2% per year develop a GI complication severe enough to require medical attention, usually a bleeding gastric ulcer. Yet even 2% of a million is 20,000 events. It is possible that these patients represent a subset of individuals with a predisposition (increased sensitivity) to greater loss of their physiologic mucosal defense mechanisms. The risk of GI bleeding with NSAID use increases with age, duration of use, comorbidities, anticoagulant use (including aspirin that may also cause the damage itself), and a history of bleeding ulcers. However, it also causes bleeding in infants.28 A subset of patients (about one-third in one series20) may suffer a modest mucosal injury that results in one of the characteristic chronic changes of reactive gastropathy. However, in a series looking at the protective use of PPIs on naproxen 500 mg b.i.d.,4 within a week 25% developed antral ulcers, 12.5% duodenal ulcers, and 9.4% ulcers in multiple locations (one developed ulcers in the antrum and body; two in the antrum and duodenum). Of those taking PPIs, only 11.8% developed ulcers, all of which were antral. Anecdotally we have seen inflammatory masses in the cardia and proximal duodenal that seem likely NSAID related. They may take weeks/months to resolve or persist for months.
The gastric mucosa of the majority of users may therefore never develop changes that can be detected by endoscopic or histopathologic examination. In practice, however, most NSAID users have been taking them for long periods of time, and it is less clear how well the stomach is able to adapt to chronic NSAID ingestion. Overall, only a small subset of chronic NSAID users have biopsies with all features we commonly associate with chemical gastropathy; most may only have foveolar hyperplasia.20 In addition, such changes may occasionally occur in persons with no history of chemical injury. Concurrent H. pylori infection makes a firm diagnosis of chemical gastropathy extremely arduous.20
Figure 13-9. A: Biopsy specimen of a subepithelial hemorrhage in a patient with alcoholism. There is diffuse subepithelial hemorrhage across the full span of the fundic gland mucosa, but there is no inflammation present. B: Mucosal edema with an empty appearance of the interpit regions throughout the span of the biopsy (arrows). This is from a biopsy specimen adjacent to an area of subepithelial hemorrhage in a patient with alcoholism.
Alcoholic gastropathy. The term alcoholic gastritis (or gastropathy) is commonly used in a clinical or endoscopic context to explain abdominal pain or gastric lesions in alcoholic patients. Gastric hemorrhages, erosions, or both are found in 20% or less of actively drinking alcoholics with GI bleeding27 (Fig. 13-9A). In humans, there are few data concerning the histologic basis of gastric erosions or subepithelial (lamina propria, rarely with submucosal) hemorrhages. However, in 1954 (pre-Helicobacter days), servicemen had their gastric mucosa examined after acute alcoholic ingestion.29 In most, a variety of lesions were noted: patchy hyperemia, erosions, petechiae, and “exudate.” Biopsy specimens showed mainly superficial gastritis with prominent neutrophils.29 Some specimens exhibited edema of the foveolar region. More recently, actively drinking alcoholics had biopsy specimens taken from either subepithelial (lamina propria) hemorrhages or erosions, with specimens for comparison from adjacent sites.27 The subepithelial hemorrhage specimens revealed foveolar region hemorrhage in target lesions and sometimes striking edema in the adjacent mucosa (Fig. 13-9B). The erosions were verified histologically in 70% and commonly exhibited a pseudomembranous appearance. In both the erosions and the hemorrhages, the associated inflammatory change was mild and was similar in severity in the lesions and the adjacent mucosa. In actively drinking alcoholic patients, the gastric mucosa may, in addition, exhibit the features of congestive gastropathy if there is portal hypertension. This is discussed in the next section.
Caustic-induced injury. Accidental or suicidal ingestion of acids or alkalis (commonly in the form of household cleaners) may cause a wide range of oral, esophageal, and gastric lesions.30, 31 The gastric antrum is especially vulnerable, with lesions ranging from superficial erosions to gangrene. A late complication in some cases is the development of gastric antral strictures.31
Graft versus host disease. is discussed in detail in Chapter 3. GVHD is seen in severely immunosuppressed patients after allogeneic bone marrow transplant where the donor T cells attack host cells leading to cell necrosis. Upper endoscopic examination in the context of suspected or proven GVHD is done if upper GI symptoms are prominent. The severity of change in the upper gastrointestinal tract (UGT) frequently do not parallel the colonic changes (see Chapter 3). The endoscopic spectrum ranges from normal to subtle swelling to erosions, ulcers, and mucosal sloughing. In addition to mild reactive changes, variable degrees of epithelial injury are seen in a background of few inflammatory cells. In general, the histopathology is that of epithelial injury/death at variance with the amount of inflammation present. In the acute phase, epithelial injury can be seen as increased apoptosis (occasionally more numerous in the neck area), attenuated regenerative-appearing epithelial cells in less injured glands, granular eosinophilic debris intermixed occasionally with nuclear debris within dilated glands, sloughed mucosa, and total destruction of gastric glands in severely injured glands. The histopathology in chronic cases can include crypt loss, inflammatory polyps in severe disease, and architectural distortion with crypt branching and atrophy.32, 33, 34 Telangiectatic vessels suggestive of gastric vascular ectasia have been identified.34 Biopsy specimens are commonly taken to also rule out infections such as cytomegalovirus (CMV). Nonetheless, similar histopathology can be seen in CMV and human immunodeficiency virus (HIV) infection, transplant recipients, and in primary immunodeficiency.34
Figure 13-10. A: With chemotherapy, gastric injury is not uniform. There is architectural distortion and individual crypts are in various stages of repair. B: Individual crypts vary from some that are very attenuated and undergoing restitution (blue arrow) to others in which more typical regenerative changes can be found (red arrows). These are irregularly admixed with more normal-appearing pits.
Chemotherapy and radiation. can cause both gastritis and stomach ulcers. The pathology is similar to that seen in chemical/reactive gastropathy with glandular atypia and increased apoptosis (apoptotic gastropathy), and there may be abnormal mitosis.35 A typical feature, identifiable at low power, is that adjacent pits with relatively normal epithelium, and pits with attenuated mucosa, and all stages between, can be immediately adjacent to each other (Fig. 13-10). Gastric injury induced in short-term exposure is often temporary. The acute response to massive irradiation occurs largely in the antrum and the prepyloric region. Not uncommonly, gastric erosions or discrete ulcers are encountered in patients with malignancy who have received abdominal irradiation and are on chemotherapy. Biopsy specimens and cultures may be obtained to rule out recurrent or metastatic disease and opportunistic infection. With larger doses, the damage may be irreversible with destruction of acidproducing glands.
Ischemia. Ischemic disease of the stomach is extremely rare. See Chapter 2 for a more detailed discussion. Atheromatous embolization of cholesterol,36 therapeutic embolization to help control bleeding, accidental entry of selective intra-arterial radiotherapy (SIRT) beads, and vasculitis37, 38 or hypovolemic states are reported causes of erosive gastritis and gastric ulcers. There have also been isolated reports of patients with chronic gastric ulcers and erosions that healed after intestinal revascularization.39 The reported histology in these cases may lack the classic features of ischemia. In severe disease, epithelial and glandular cells are shed in the lumens of pits. Although this sounds innocuous, the mucous-producing cells can take on the appearances of signet ring cells, mimicking signet ring carcinoma (Fig. 13-11), analogous to similar lesions seen in pseudomembranous colitis. Another potential mimic of signet ring cells are the normal mucous-producing cells that appear in the oxyntic mucosa as it approaches the antrum. The polarity of these cells may appear abnormal, but these are terminally differentiated cells with no proliferative activity (Fig. 13-11E,F), while parietal cells can be shed into the lumen in oxyntic mucosa and raise the question of parietal cell carcinoma because of apparent disorderly sheets of cells (Fig. 13-12). The gastroduodenal subepithelial hemorrhages and erosions reported in some children with Henoch-Schonlein purpura might be due to vasculitis-induced mucosal ischemia.40
Figure 13-10.(Continued)C: Overview of second biopsy with more severe changes. The admixture of pits of different stages of degeneration and repair, some benign columnar and others lined by restituting epithelium. D: Severe chemotherapy changes with most glands being lined by restituting epithelium although focally they are more columnar.
Predominantly Vascular Changes
Gastric antral vascular ectasia. is an uncommon cause of chronic GI bleeding with occult iron deficiency anemia. It is characterized by telangiectatic capillaries with fibrin thrombi and marked fibromuscular hyperplasia of the lamina propria41 (Fig. 13-13). Though it has been primarily described in the gastric antrum, proximal involvement has been reported.42, 43 Vascular ectasia can be seen in a number of other conditions, including portal hypertension,44 endstage renal disease,45 and congestive heart failure.46 Histology is rarely needed to confirm the diagnosis when the endoscopy has a characteristic watermelon appearance; however, sometimes differentiation from portal hypertensive gastropathy can be problematic (see Chapter 2).47
Figure 13-11. Ischemic change with signet ring cells. A: Active erosion with fibrinous exudes and hemorrhagic ischemic change. B: “Pseudomembranous gastritis”-like appearance with “signet ring” cells (SRCs) on the surface.
Figure 13-11.(Continued)C: Numerous SRCs observed in and out of degenerated glands on ischemic background. D: SRCs with abundant cytoplasm and compressed nuclei, mimicking infiltrative adenocarcinoma but almost too monomorphous for carcinoma. E: A further possible mimic of signet ring cells are the mucous cells normally found in the superficial midzone of the oxyntic mucosa toward the antrum. F: The mib-1 shows this zone to have no proliferative activity.
Figure 13-12. Gastric ischemia. A: The deep specialized compartment of the mucosa is ill-defined, lacking a crisp definition seen superficially. B: Detail reveals that the specialized glands have been shed into the lumen of the pits and that PCs are particularly conspicuous. The patient also had contiguous infarction of the small intestine. (Courtesy of Dr. R. Barr.)
Figure 13-13. A: GAVE with numerous dilated capillaries and intervening muscle in the lamina propria, here quite marked, interweaving between the pits. B: GAVE with a thrombosed vessel in the lamina propria (bottom left) and marked reactive epithelium, which is seen frequently in this condition. C: Bleeding from GAVE.
Portal hypertension (congestive gastropathy). In cirrhosis, especially when accompanied by portal hypertension, a number of endoscopic abnormalities have been described; they are thought to represent the consequences of portal hypertension and mucosal congestion.48 These are considered here because sometimes they are associated with discrete subepithelial hemorrhages. In the fundus and proximal body there may be a mosaic pattern, sometimes described as a snakeskin appearance. These are red areas separated by fine white serpentine reticulations.48 Another appearance is that of discrete red spots, especially in the antrum.48 Biopsy specimens may reveal focal mucosal vascular ectasia49 that can be mistaken for chronic inflammation to the unwary (Fig. 13-14), but the ectasia may be much more striking in the submucosa50 deep to the zone usually sampled in endoscopic biopsies50 (see Chapter 2).
Hemorrhagic gastropathy (“gastritis”) and “Curling’s ulcer”. Stress/shock/burn-related ulcer is the most serious form of gastropathy that usually occurs in critically ill patients, including organ system failure, sepsis, burns, and intracranial disease.51, 52 The term “gastritis” may have a degree of truth historically in that many of these patients may well have had concomitant Helicobacter gastritis, but it is primarily a gastropathy.
This condition is usually a complication of severe trauma leading to profound physiologic stress with hypovolemia or hypoxia (as in shock), but can also be seen in binge drinking alcoholics. It used to be common post surgery, but with better intensive care and prophylactic antisecretory therapy, this is now rare. In shock and other low-flow conditions, mesenteric blood flow is shunted to the systemic circulation to maintain perfusion. Shunting mesenteric blood flow to the systemic circulation is often accomplished at the expense of adequate blood flow to the mucosa, with back perfusion of acid into the lamina propria and subsequent hemorrhage. Such shunting is a major contributing factor to stress-related mucosal disease and acute hemorrhagic gastritis.
Figure 13-14. Portal hypertensive gastropathy. A: The overview suggest chronic gastritis. However, detail (B) suggests that these are all small endothelial-lined channels with small lumina. C: CD31 immunostain shows that these are endothelial. D: Endoscopic appearances of portal gastropathy. Note not only the varices but the numerous telangiectases in the background that are the endoscopic counterpart of the biopsy.
The pathology is inconspicuous except for areas of mucosal hemorrhage that can precipitate significant acute blood loss. In pre-PPI days this condition was often fatal and involved the entire stomach. Early lesions seem to predominate in the fundus and more proximal body; later there is more distal spread to involve the antrum. It is very uncommon to have only antral involvement. Stress lesions are usually superficial. Areas of hemorrhage are seen at the tips of the mucosal folds down to the mucous neck region.53, 54, 55, 56 Hemorrhagic gastropathy, which frequently occurs in the early postburn period and is often manifested by coffee-ground emesis, damages the mucosa, rendering it more susceptible to ulceration “Curling’s ulcer.”57 Bleeding from hemorrhagic gastropathy is commonly trivial and ceases with resumption of GI function. GI bleeding later in the postburn period should suggest the diagnosis of Curling’s ulcer.
Curling’s stress ulcers of the stomach or duodenum were once the most frequent life-threatening GI complications of burn patients. These are usually diagnosed at operation or at autopsy after the 3rd postburn day in patients with larger burns. These stress ulcers are usually preceded by hemorrhagic gastropathy (“gastritis”), and often result in hemorrhage and perforation more often than intestinal ulceration58—thus, they have correspondingly high mortality rates.57 The limited literature on Curling’s ulcers describe engorged submucosal capillaries, a paucity of inflammatory cells, and a sharp demarcation of necrotic mucosa showing cellular degeneration from viable apparently uninvolved mucosa.57 The deeper lesions usually contain a base of necrotic debris without the scarring or well-formed granulation tissue seen in ordinary gastric ulcers.
The apparent decrease in major complications (bleeding and perforation) may be more due to improved cardiorespiratory and nutritional support53 than to routine acid-neutralizing or acid-inhibiting prophylactic therapy.59 Endoscopic studies have shown that the lesions develop within hours of the illness or trauma.54, 55, 56, 60 If the underlying disease is reversed, the lesions vanish in parallel. It is unlikely to receive a biopsy in this condition but it can be in the differential in a gastrectomy specimen of a patient with hypovolemia secondary to severe hemorrhage. Patients with stress/shock ulcer often are given continuous PPIs that are also given prophylactically to patients at risk before surgery.
Of interest, gastric mucosal damage seen in marathon runners can also be explained by shunting of mesenteric blood flow to the periphery.61, 62, 63, 64 Gastric erosions and ulcers in marathon runners are mainly localized in the corpus,61, 62, 63 where stress or shockassociated gastritis is normally seen.64 Mucosal erosions can also be seen in the antrum (sometimes primarily), suggesting altered gastric physiology and microcirculation induced by intense exercise.65, 66
Distinctive (Specific) Types of Gastritis
The designation distinctive or specific refers to histologic features that markedly narrow the differential diagnosis or are occasionally pathognomonic. We subclassify these into infectious and noninfectious (Table 13-4).
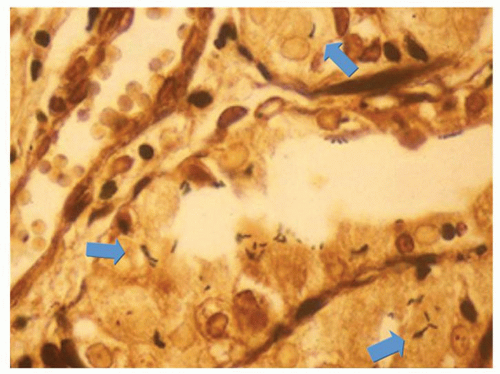
Infections. The most common infection in immune competent patients is H. pylori infection. It has an acute phase as demonstrated by volunteers swallowing the organism, and then most pass on to a phase of chronic relatively asymptomatic infection. Although many organisms have urease and hence can potentially survive in the stomach for short periods, there are few data to suggest that other bacteria cause an acute infectious gastritis rather than what is euphemistically called infectious gastroenteritis; however, a patient with enterococcal gastritis has been described presenting with “dyspepsia”67 (Fig. 13-15). These organisms are readily visible on an hematoxylin and eosin (H&E) stain but any bacterial stain can enhance their visibility. They are Gram positive. The challenge is not to interpret them as coccal forms of Helicobacter, which are not overtly adherent to the surface epithelium (they are dead), or as incidental organisms of oral or duodenal origin that can grow in the stomach in patients with marked hypo or achlorhydria, whether pathologic or iatrogenic (PPIs). They are negative with Helicobacter immunohistochemistry.
Amongst viral infections, CMV is the most common in the stomach and is found especially in children. How long the infection needs to be present before becoming symptomatic, and therefore whether it is really acute, is unclear. It is also the cause of childhood large gastric folds—pediatric “Ménétrier’s disease.” Apparent incidental inclusions are occasionally found, primarily in epithelial cells, and are thought to represent a carrier state. In immunodeficient patients, whether iatrogenic, acquired, or congenital, a variety of infections including CMV may be encountered (Fig. 13-16, Chapter 19, and subsequently in this chapter—Ménétrier’s disease). General viremias may be associated with a variety of other viruses that may be found incidentally such as measles (Fig. 13-17), herpes, and Parvovirus infection.
AH. pyloriinfection.Helicobacter pylori is etiologically linked to histologic gastritis, PUD, marginal zone lymphoma, and gastric carcinoma in addition to other conditions (Table 13-5). The discovery of Campylobacter pylori by Warren and Marshall in 1982 and subsequently renamed Helicobacter pylori, was preceded by nearly 100 years of inconspicuous publications relating spiral bacteria to achlorhydria, gastritis, gastric urease, and antimicrobial therapy for ulcers.68, 69, 70, 71 Koch’s postulates for H. pylori were fulfilled by volunteers who ingested the organism and produced gastritis, one self-limited9 and the others more persistent.72 In 1984, the bacterium was cultured from 88% of patients with gastritis and the bacteria was not cultured from any patient with histologically normal gastric mucosa.73 Furthermore, the inflammation associated with H. pylori gastritis is reduced with antibiotics that suppress H. pylori.74, 75 This direct evidence of a causative role was complemented by indirect evidence that H. pylori may have been responsible for two miniepidemics of nonerosive, “nonspecific” gastritis.76, 77 These miniepidemics were reported in volunteers taking part in secretory studies. The individuals developed self-limited upper abdominal complaints, with hypochlorhydria that continued for months in some. In this “epidemic gastritis,” the pattern of inflammation was a superficial gastritis, suggesting severe functional impairment of parietal cells rather than their obliteration. In 2005, Barry Marshall and Robin Warren jointly received the Nobel Prize in physiology and medicine for their “discovery of the bacterium Helicobacter pylori and its role in gastritis and peptic ulcer disease”.
Figure 13-15. Enterococcal gastritis. It is important not to interpret these as coccal forms of Helicobacter, which never exist in the absence of typical forms of Helicobacter as they are not viable. (Triple stain with carbol fuchsin.)
Figure 13-16. CMV gastritis with ulceration and perforation. This occurred in a patient with Crohn’s disease on steroids. A: Perforation—the mucosa is re-epithelializing the ulcer that goes through the muscularis propria. B: Mucosa with chronic inflammation and numerous CMV inclusions, (C) also demonstrated immunohistochemically.
Figure 13-17. Measles in a gastric biopsy. Numerous Warthin-Finkeldey giant cells typical of measles can be seen. A specific antimeasles antibody was used to confirm the diagnosis immunohistochemically (right). The patient was prodromal when the biopsies were taken and developed a typical rash soon after. (Courtesy of Dr. M Vieth, Bayreuth.)
EpidemiologyHelicobacter pylori infection almost certainly is the most common chronic bacterial infection in humans and is present in approximately 60% of the world population.78Helicobacter pylori gastritis is for the most part acquired before age 10.79 The prevalence of H. pylori infection varies both between and within countries.80 This relates to the known determinants of infection, particularly socioeconomic standards of living of the young.81, 82, 83, 84, 85 Differences in prevalence among ethnic groups of similar socioeconomic status reflect differences in the environment and possible host genetics.82, 86 In countries where there has been rapid economic development with associated improvements in standards of living, there is some evidence that the prevalence of infection is declining.87, 88 In developed countries, it is currently uncommon to find infected children, but there is a cohort effect so that the percentage of infected people increases with age, being about 50% in those over the age of 60.89, 90, 91 The higher prevalence among the elderly reflects higher infection rates when they were children rather than infection at later ages. Nonetheless, H. pylori infection remains common among the socially disadvantaged and in the large immigrant population in developed countries.92, 93
Table 13-5 AHelicobacter-associated Diseases
Gastric conditions associated with H. pylori infection
Gastritis
Duodenal ulcer
Gastric ulcer
Gastric carcinoma
Gastric lymphoma
Extragastric conditions possibly associated with H. pylori infection
Although the exact route of transmission is not known, person-to-person transmission, oral-oral or fecal-oral, as exemplified by data on intrafamilial clustering is most likely.94, 95 Possibilities include the common practice of (grand)parents masticating food before feeding to infants, bearing in mind that the act of burping showers the oral cavity with gastric organisms. The role of external reservoirs in H. pylori transmission has not been ruled out, particularly in rural and developing areas.96 Nonetheless, studies on water, one of the most well-studied ecosystems, yielded inconsistent results.97, 98, 99 The inconsistent results may reflect different water treatment modalities and/or variations in polymerase chain reaction (PCR) procedures. It is important to remember that mere presence of DNA in a potential environmental reservoir is not a clear evidence of the transmissibility of the organisms, as the organisms may or may not be viable. A culture of H. pylori organisms from these sources would provide stronger evidence—H. pylori has not been cultured to date from water reservoirs, but has been found in streams, possibly the result of fecal contamination.
Most interestingly, H. pylori strains from different geographical areas exhibit clear phylogeographic features that provide information about the migration of human populations.100 Sequence differences in seven core housekeeping genes enabled grouping H. pylori into seven population types based on geographical associations (hpEurope, hpEastAsia, hpAfrica1, hpAfrica2, hpAsia2, hpNEAfrica, and hpSahul).101, 102, 103 These studies suggest H. pylori has spread from East Africa over the same time period as modern humans, around 58,000 years ago.104Helicobacter pylori has remained intimately associated with the human host populations ever since.102 Overall, H. pylori sequences can potentially provide details of human migrations that would otherwise be difficult.104
Some strains of Helicobacter are clearly associated with more inflammation, the ability to produce ulcers, atrophy, and carcinoma. The best known of these is the CagA gene that is present in 50% to 70% of H. pylori in Western societies. It resides in the CagA pathogenicity island (PAI), although a vacuolating toxin (VacA) can also be present. The CagAPAI contains about 30 genes, but is usually absent from H. pylori strains isolated from humans who are carriers of H. pylori but remain asymptomatic. The CagA gene codes for a relatively long (1,186 amino acid) protein. The CagAPAI also includes a gene coding for a complex type IV secretion system. About 50% to 70% of H. pylori strains in Western countries carry the cag PAI.105 Western patients infected with strains carrying the cag PAI have a stronger inflammatory response in the stomach and are at a greater risk of developing peptic ulcers or stomach cancer than those infected with strains lacking the island.106
Following attachment of H. pylori to gastric epithelial cells, the type IV secretion system “injects” a pro-inflammatory peptidoglycan from the organisms’ own cell wall into the epithelial cells. The injected peptidoglycan is recognized by NOD1, which is a cytoplasmic pattern recognition receptor (immune sensor), which then stimulates expression of proinflammatory cytokines.107 The type IV secretion system also injects CagA into the epithelial cells, where it disrupts the cytoskeleton, adherence to adjacent cells, cell polarity, and intracellular signaling.108 Once inside the cell, the CagA protein is phosphorylated on tyrosine residues by a host cell membrane tyrosine kinase. There may also be activation of the epidermal growth factor receptor (EGFR), a membrane protein that also has a tyrosine kinase domain, and this activation is associated with altered signal transduction and gene expression in host epithelial cells, all of which likely contribute to pathogenesis. A C-terminal region of the CagA protein can also regulate host cell gene transcription independent of protein tyrosine phosphorylation.109
H. Pylori-associated GastritisHelicobacter pylori gastritis is for the most part acquired before age 10.79 Today, many believe the hypochlorhydria accompanying febrile childhood infections predisposes children to H. pylori infection, as hypo or achlorhydria often develops during and after acute infectious diseases such as influenza, tonsillitis, pneumonia, and bronchitis. In several cases, in spite of normal acid secretion before the disease, lowered secretion was documented for a long time after the disease. It is unclear if these changes are secondary to direct bacterial involvement or due to bacterial toxins as acute gastritis with complete achlorhydria can be produced following intravenous injection of dogs with diphtheria toxin. Also, a fall of secretion, amounting at times to achylia, is seen in toxemias of pregnancy, especially eclampsia.110
Disease outcome varies from no symptoms in many patients, to duodenal ulcers, gastric ulcers, gastric mucosa-associated lymphoid tissue (MALT) lymphoma (marginal zone lymphoma), and gastric carcinoma in others. While host and environmental factors play critical roles in disease outcome, it is well known that the development of a specific disease is associated with a specific gastritis pattern111, 112 (see Fig. 13-1). DU is typically associated with antral-predominant gastritis, little or no oxyntic gland (corpus and fundus) atrophy, and normal or increased acid secretion.113, 114, 115, 116 Gastric ulcer and the intestinal type of gastric cancer are typically associated with pangastritis, widespread oxyntic atrophy with varying degrees of intestinal metaplasia, and hypo- or achlorhydria,113, 117, 118, 119 (see Fig. 13-1).
The distribution and severity of H. pylori-related gastritis (and thus disease risk) is related to the distribution and density of H. pylori within the stomach.120 The distribution of H. pylori within the stomach is influenced by a person’s acid secretory status.120, 121, 122Helicobacter pylori are well adapted to the human stomach with its acidic environment that is hostile for most microorganisms by producing large amounts of the enzyme urease. Urease catalyzes hydrolysis of urea present in the stomach to yield ammonium ions () and CO2 in a thin neutral layer around the outer surface of the bacteria.123 The ammonium ions markedly increase the pH in its surrounding environment to neutral (or above), which is necessary for its survival.124Helicobacter pylori further protects itself by swimming through the protective layer of gastric mucus away from the acidic contents of the lumen toward the more neutral pH environment of the epithelial cells beneath.125 Nonetheless, H. pylori flourishes best at a pH range of 3.5 to 5126 where in the presence of urea it can maintain a proton motive force across its periplasmic membrane, ensuring a continued supply of energy through ATP synthesis.126 In a high acid output microenvironment, this protective mechanism cannot keep up with the high hydrogen ion influx and the bacterium dies.126, 127 This explains why early H. pylori gastritis is concentrated in the antrum, although the organism may also be able to produce a protein that reduces gastric acidity, facilitating colonization.
Similarly, when the microenvironment pH rises above about 5, the bacteria produces ammonia in excess of what is needed to neutralize the muchdiminished influx of hydrogen ion. The microenvironment then becomes increasingly alkaline, which is also detrimental to the bacteria. If Helicobacter pylori‘s continual production of ammonia raises the pH above 8, the bacteria cannot survive.126, 127, 128, 129, 130 This explains why H. pylori may not be identified in achlorhydric states (gastric atrophy and with continued PPI use) when the inflammatory pattern is clearly that associated with low H. pylori density. Further, in patients on PPIs the organism may migrate to the oxyntic mucosa to find enough acid to survive, hence the frequently observed proximal migration of Helicobacter with both increasing atrophy and PPI use. Not only that, but the organisms seem to migrate deeper into the oxyntic glands as opposed to their usual superficial location, and can even be found within canaliculi of parietal cells.
Histology ofH. pylori-associated gastritis. The histologic spectrum of H. pylori-associated gastritis ranges from minimal to severe inflammation. When present, neutrophils are concentrated in the pit regions; uncommonly, they infiltrate the surface in a dense fashion, often associated with erosions. The association with neutrophils has been stressed, in part because they are not normally present in the lamina propria. Thus, their presence indicates inflammation and active disease, whereas with mononuclear cells, an increase is much more difficult to appreciate unless quite marked. Occasional chronic inflammatory cells are acceptable as part of the normal.131 In H. pylori-associated active gastritis, the number of mononuclear cells is also increased.132 The inflammation is worse along the lesser curve than on the greater curve with the most severe inflammation at the antral-body transitional mucosa. Lymphoid hyperplasia (Fig. 13-18) with numerous lymphoid follicles is seen in some cases, especially in children.133, 134 Similar to mononuclear cells, unorganized lymphoid aggregate abutting the muscularis mucosae is normal.131
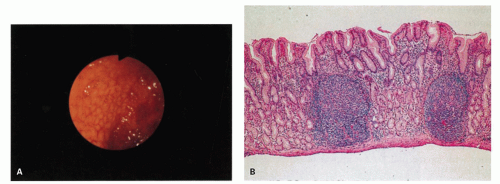
Figure 13-18.Helicobacter pylori in a child. A: Pronounced antral gland nodularity at endoscopy. B: Prominent lymphoid follicles and otherwise mild inflammation are revealed in this H. pylori-positive biopsy specimen. (Courtesy of Drs. E. Hassall and J. Dimmick.)
The epithelial mucin ranges from intact to markedly depleted (reactive changes), with mild depletion being the most common pattern. With progressive atrophic gastritis, especially when accompanied by intestinal metaplasia, there is a reduced frequency of association with H. pylori.135 Indeed, it is rare to find Helicobacter when intestinal metaplasia is present.
Helicobacter pylori may be present in biopsy specimens that are histologically close to normal, especially in the gastric body132, 136, 137 (Fig. 13-19), and in patients with much more active disease in the gastric antrum that may not have been well-sampled. In such examples, the mild inflammation is limited to the superficial lamina propria, especially in the oxyntic mucosa where it is easily overlooked on a cursory examination. This also tends to occur with Helicobacter species other than H. pylori. However, we have never seen an absolutely normal (noninflamed) mucosa in an infected patient if biopsies from both antral and oxyntic mucosa have been taken. This becomes important practically.
Figure 13-19. Our current minimal inflammation in a patient with H. pylori gastritis. Antral mucosa is in the left three panels and oxyntic mucosa in the right three. Appreciating this is important in laboratories in which some form of routine stain is not employed, and the pathologist relies on the quantity of inflammation to justify ordering a special stain or immunostain. The lower two panels are anti-Helicobacter immunostains at both sites. The numbers of organisms in both are readily appreciated.
1. When routine special stains for Helicobacter come with gastric biopsies, some meticulously examine them until they realize the time has been spent to no purpose, so they make a decision as to the minimal amount of inflammation required to seriously examine the special stain, and when they can rest assured that no organisms are present. In all likelihood absolutely normal (non-inflamed) biopsies need minimal examination of special stains provided. As shown in Figure 13-19 when Helicobacter are present in minimally inflamed biopsies organisms are invariably numerous so readily detected. They are also likely to be non-toxin producers and have virtually no pathogenic potential, as complications follow organisms producing considerable inflammation.
2. When no special stains are carried out routinely on all gastric biopsies, a decision has to be made on the minimal amount of inflammation to trigger this (Fig. 13-19).
3. If the special stain routinely carried out in a lab is a stain in which one cannot have real confidence (e.g., Giemsa stain), a backup is required (e.g., silver stain or an immunostain) in which one does have more confidence is required when inflammation equal to or more than that shown in Figure 13-29 is present, and organisms are not identified. Especially when one is convinced that the organism may be present.
4. When patients have recently undergone eradication therapy but still have chronic inflammation quantitatively compatible with Helicobacter being present, we usually revert to a silver or immunostain before declaring that “Helicobacter are not identified”.
When organisms are present, especially if scant, although it seems mundane, it really is necessary to ensure that the organism is H. pylori or other Helicobacter species. Although therapy sounds easy, there is a risk of complications such as the NAP1 strain of Clostridium difficile, which can be potentially lethal. Our approach is that if organisms considered to be Helicobacter are identified, you should be able to photograph it and others will “buy it.” We do not make the diagnosis on coccal forms unless accompanied by an immunostain with regular organisms as they are otherwise nonviable and easily mistaken for other cocci— discussed subsequently. Most biopsy specimens with diffuse chronic active gastritis are associated with H. pylori. Sampling error because of the patchy distribution of the organisms, and especially failure to biopsy both antral and oxyntic mucosa, may be one of the reasons that a minority of cases of diffuse chronic gastritis lack H. pylori.136 However, recent use of antibiotics, PPIs, recent eradication therapy, atrophy and intestinal metaplasia, and other rare causes such as diffuse chronic gastritis are all reasons organisms may not be found.
ACUTE PHASE: The initial, acute phase of infection is subclinical in the great majority of subjects. As in any acute infection, there is lamina propria edema with neutrophilic infiltration of foveolar and surface epithelium—acute neutrophilic gastritis. Acute H. pylori infection can temporarily induce a period of hypochlorhydria, which probably facilitates widespread colonization and a pangastritis. In a small minority of people, and particularly in childhood, the organisms may be spontaneously cleared,138 the polymorph infiltrate resolves, and appearances return to normal. Nonetheless, the acute phase is short-lived. Of those infected, the majority fail to eliminate the infection. Over a couple of weeks, there is a gradual accumulation of chronic inflammatory cells that come to dominate the histologic picture131—chronic active gastritis.131, 139 It may take weeks or months for acid output to return close to preinfection levels, and in a proportion of patients output remains low. In general, as the hypochlorhydria seen in the acute phase of infection is short-lived, early H. pylori gastritis is typically antrum-predominant.
EARLY H. PLYORI INFECTION: H. pylori colonization is usually accompanied by inflammation.
This involves secretion of a protein that decreases acid secretion, allowing colonization, potent neutrophil chemokines, and mast cell degranulation that increases polymorph immigration.140, 141, 142Helicobacter pylori density, and the presence of a PAI (see previous discussion), influence the severity of inflammation and subsequent damage over the course of lifelong infection.
Early H. pylori gastritis is typically antral predominant where gastric acidity is reduced by antral mucin. The antrum shows diffuse superficial or full-thickness infiltration by lymphocytes, eosinophils and plasma cells, occasional lymphoid follicles, and variable infiltration with neutrophils within the lamina propria but more commonly infiltrating the foveolar and surface epithelium. In early H. pylori gastritis, inflammation in the gastric corpus is mild, superficial, or even absent120, 143 (Fig. 13-20A,B). Inflammatory sequelae, such as intestinal metaplasia, are for the most part (initially) confined to the antrum or angulus.
Early H. pylori gastritis is often characterized by an exaggerated gastrin response to meals and other stimuli,144 which precipitates an increase in acid secretion enough to cause duodenal ulcer disease in some patients. A consistent histologic finding associated with marked duodenitis occurring in some patients with early H. pylori gastritis is surface gastric metaplasia—patches of gastric type mucous cells interspersed between the absorptive and goblet cells of the duodenal epithelium. Helicobacter pylori, when present in the duodenum, can only be identified in areas with gastric metaplasia. It is hypothesized that in the duodenum, similar to the gastric mucosa proper, H. pylori induces inflammation and erosions where it resides—gastric metaplasia (and heterotopia) patches,145 although in our experience this has a low yield (see subsequent section on duodenal disease). As such, the prevalence of duodenal gastric metaplasia is much lower in “healthy” volunteers.145
Figure 13-20. Stages in the natural history of H. pylori. Biopsies from the antrum are on the left and the oxyntic mucosa on the right. A,B: Initial infection affects the antrum with minimal involvement of the oxyntic mucosa. C,D: Over time this extends to the oxyntic mucosa so that there is superficial inflammation but no atrophy. E,F: Ultimately the chronic inflammation extends into the specialized mucosa with gland loss. (This can also be exaggerated or mimicked with long-term PPI use.)
Figure 13-20.(Continued)G,H: Finally the oxyntic mucosa becomes lost completely and is replaced by pseudopyloric metaplasia, as shown here (invariably with endocrine cell hyperplasia), or with intestinal mucosa. The inflammation in the antral mucosa varies from virtually none when there is severe hypochlorhydria to modest if there is still sufficient acid secretion to support antral organisms (which require acid to prevent their urease causing too alkaline an environment).
ADVANCED H. PYLORI INFECTION: With sustained H. pylori infection, there is a gradual reduction of acid-producing mucosa because of gradual proximal spread of inflammation that facilitates H. pylori‘s proximal migration. Helicobacter pylori colonization is accompanied by the inflammation. As there is a gradual reduction in the acid-producing mucosa,146, 147, 148 the inflammatory front advances proximally with disease progression (Fig. 13-20B). The development of hypochlorhydria and even achlorhydria, including use of PPIs, facilitates proximal migration of the bacteria, which allows the development of corpus gastritis, and eventually corpus atrophy (Fig. 13-20C,D). This is the setting in which gastric ulcer and later gastric carcinoma develops (see Fig. 13-1).
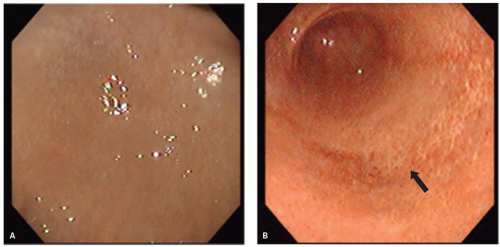
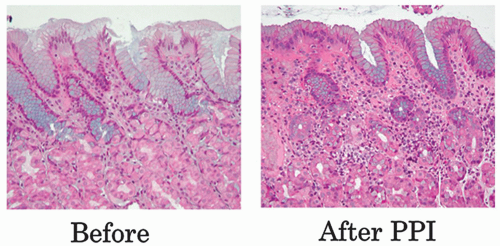
The natural history of H. pylori gastritis is for the inflammation to progress diffusely from the antrum into the adjacent corpus resulting in an atrophic front of advancing corpus injury (that may be visible endoscopically to the trained eye—Fig. 13-21), leading to a reduction in acid secretion and eventually loss of parietal cells and development of corpus atrophy.120, 149, 150 The front progresses uniformly and so appears to advance faster on the lesser curve (Fig. 13-22). This scenario is accelerated in clinical situations associated with low acid secretion such as chronic therapy with PPIs—widely used in gastroesophageal reflux disease (GERD)151, 152, 153 (Fig. 13-23). Thus, antral-predominant gastritis may in some instances represent an earlier stage of atrophic pangastritis, these patterns representing two ends of the spectrum of “H. pylori infection” rather than mutually exclusive diseases.14, 120, 154
H. pyloridiagnosis. Depending on endoscopy need, diagnostic testing for H. pylori can be divided into invasive and noninvasive methods.155 Noninvasive methods do not require endoscopy and include serology and urea breath test. Invasive tests require endoscopy; this group includes tests for urease, histology, and culture. The choice of test depends on the clinical situation (e.g., patient requires evaluation with upper endoscopy) and on other issues such as cost, availability, population prevalence of infection, pretest probability of infection, and factors such as the use of PPIs and antibiotics, which may influence certain test results.
Noninvasive methods
1. Serology: Antibodies to IgG and IgA have been used successfully to study the epidemiology of H. pylori in different populations in different parts of the world. However, serology remains positive long after successful treatment of the infection and cannot be used to assess treatment outcome. Serologic tests are often species specific (H. pylori) and can be negative with infection with other Helicobacter species, for example, Helicobacter heilmannii. In addition, inaccurate tests are also more common in the elderly and in patients with cirrhosis in whom specificity can be compromised. As a result, other techniques are preferred in these settings.156, 157, 158
Figure 13-21. Left panel shows the natural and indistinct transition from antral oxyntic mucosa. That on the right shows the atrophic front (arrow). Prior to the rediscovery of H. pylori, this was thought to be an aging change. (Courtesy of Dr. Taiji Akamatsu.)
Figure 13-22. Advancing gastritis with time. “A” depicts Helicobacter-inflamed antrum and “B” Helicobacter-inflamed or possibly normal corpus/oxyntic mucosa. 1. No atrophy. 2. Atrophy is thought to begin at the junction of the antral-corpus junction and spreads proximally in a cone-like manner. Often this equates with the angulus but the histologic junction is variable. 3. With time the cone extends proximally, but 4. Because the lesser curve is much shorter than the greater curve, the lesser curve is completely atrophic long before the greater curve and fundus. It is therefore possible to have biopsies from the lesser curve with complete atrophy and metaplasia but relatively normal oxyntic mucosa on the greater curve.
2.Urea breath tests, with 13C or more widely available14C, provide a powerful, noninvasive tool for research. The principle is that the carbon-labeled urea is hydrolyzed by the urease in the H. pylori, if present, resulting in the formation of ammonia and carbon dioxide. The amount of labeled carbon dioxide in the breath is then measured.159 The urea breath test provides an accurate assessment of H. pylori status that rivals histology for being the gold standard,159 but can be negative in cases with a very low level of infection.
3.Stool antigen: The presence of H. pylori in the stool of infected patients has led to the development of fecal assays.160, 161, 162 The stool assay shares a limitation of tests that use urease as a marker for the organism.163, 164 Though this is a noninvasive method, any of us who has been asked for a stool specimen might sympathize with its difficulty.
Invasive methods
1.Rapid urease test is based on the organism’s urease activity. It can be used at the “bedside” in the endoscopy unit. A gastric biopsy specimen is placed in contact with a pellet or solution that contains urea and a pH color indicator. The color changes when the pH rises above 6.0 as a result of hydrolysis of urea to ammonia.165
2.Culture: Although culture is the theoretical gold standard as there is an excellent correlation with histologic identification, in practice and in many research studies, histologic identification is used as the gold standard. One reason is that in many laboratories, cultures are less frequently positive than histology and serology. This may be due to the fact that small numbers of organisms cannot always be cultured or identified, and also because there is varying expertise in different laboratories for culturing these organisms. In general, H. pylori is a fastidious and slow-growing organism that is difficult to culture.166
Figure 13-23. Oxyntic mucosa before and after a 2-week course of PPIs in a Helicobacter-positive patient. The increase in chronic inflammation over this time frame is readily apparent.
3.Histology is currently considered the “gold standard” for detecting the infection for both untreated individuals and following therapy. The advantages of histology include the ability to document H. pylori infection, the degree of inflammation, and any associated pathology such as intestinal metaplasia, cancer, or lymphoma. The detection of organisms is enhanced by using special staining techniques discussed subsequently with their pros and cons in different clinical scenarios (see Table 13-9).
What are we looking for and where?Helicobacter pylori is a small (3 × 0.5 µm), gram-negative, wavy rod-shaped bacteria. The organisms can be visualized on the luminal side of gastric surface and pit mucous cells, in or near the adherent mucous. Often the organisms seem to cluster near more normal-appearing mucous cells (Fig. 13-24) rather than near those exhibiting severe mucous depletion. A similar situation applies when H. pylori is associated with erosive gastritis. That is, the organisms do not overlie the erosion or the very reactive mucosa immediately adjacent to it; rather, they are present in adjacent, less abnormal mucosa. Also, as discussed in the subsequent section on duodenitis, H. pylori can sometimes be seen in the duodenum only in association with gastric surface cell metaplasia. Although we look for its characteristic shape, only a small proportion of organisms are in the plane of section and so have the characteristic shape.
Hematoxylin and Eosin: Few studies report the accuracy of an H&E stain,167, 168 and it varies with the type of stain used. Experienced pathologists overall will have higher accuracy than junior pathologists in evaluating H&E-stained slides.168 Even the staining intensity varies among biopsies within a given laboratory even in the same batch, and they are especially difficult to see if all are adherent to the epithelium. Nonetheless, within and between biopsy specimens there is often considerable variation in the numbers of organisms. H&E-stained slides have low sensitivity, in particular, in biopsies with low bacterial density.168 This is particularly observed in patients using PPIs, in patients who recently received antibiotics, but also if biopsies are only taken from one part of the stomach instead of both antrum and oxyntic mucosa. Typically, there is chronic inflammation, but few or rare H. pylori organisms, and minimal or no active inflammation.
It is also important to remember that studies evaluating H&E stains are not evaluating the same stain. Though H&E stain is the most widely used stain in medical diagnosis, there are a large number of H&E protocols. Primary differences are dye composition, staining protocol, and intensity of the blue dye. Staining contrast (including H. pylori) differ depending upon the approach that is used. For example, there are actually two very closely related compounds commonly referred to as eosin. Eosin Y (also known as eosin Y ws or eosin yellowish) that is the most often used, and has a slightly yellowish cast. The other eosin compound is eosin B (eosin bluish or imperial red) and has a faint bluish cast. The two dyes are interchangeable, and selection of a particular eosin (or H&E protocol) is more a matter of preference and tradition. Overall, evaluating sections stained only with H&E leads to inconsistent results and special stains have been recommended whenever inflammation is present and organisms are not readily identifiable on an H&E stain.13
Figure 13-24. A: Detail (high dry) view of H. pylori organisms at the entrance of a gastric pit. With a little experience, they can be recognized (as gray-blue) in conventionally stained sections. B: Higher-power (×600) view showing the organisms more clearly with a modified Giemsa stain.
However, if organisms are clearly identified on H&E stain in the presence of a compatible inflammatory infiltrate, this is usually adequate for diagnostic purposes. A useful rule of thumb is that, for any diagnosis of H. pylori, you have to be able to “photograph it and stand up in court and defend the diagnosis.” If this cannot be done, then a special stain is needed. Deciding when to order a special stain is itself fraught with problems, and such a “pragmatic approach” leads to underdiagnosing Helicobacter infection about 10% of the time when a decision is made on whether “sufficient” inflammation is present to justify ordering the special stain. Overall, pathologists do not appreciate how little inflammation may be seen in some biopsies with Helicobacter (see Fig. 13-19). While one could argue that if there is little inflammation, then the chances of that organism producing complications are small. This may not hold true if, for example, only antral biopsies are obtained showing only scant inflammation and no Helicobacter, as these may be ravaging the oxyntic mucosa.
Policies for Detecting Helicobacter There is no “correct” protocol for histologic diagnosis of Helicobacter, and it depends on numerous factors that can be the driving force including the following:
1. The likelihood of organisms being present. In some populations, the positive rate is 10% to 20%, and in other >80%. In some commercial labs, it may even be supplier dependent; for an endoscopy group, serving a Western-white population may have an incidence that is <10%, whereas one serving, for example, a primary immigrant population from high Helicobacter prevalence areas, may have a rate in excess of 90%.
2. Commitment to turn-around time (TAT). Even though the vast majority of biopsies really are nonurgent, if a lab is committed to, for example, a 24-hour TAT, then this becomes the driving force. and an H&E and good special stain (silver or immunostain) may be done routinely. While in some biopsies the organisms may be easily visible at a first glance, opinions on how much time should one spend on one biopsy hunting for the organisms on H&E stain may vary when these are not that readily visible. This also gets into the issues of resource utilization and individual ego, where some take extra pride in their ability to find the organisms on H&E stain only and saving money (for the patient or institution). However, if the biopsies are normal, or the organisms are overtly detectable on the H&E stain, one can raise an ethical question of justification to bill for tests that were not really required. Unfortunately while opinions are many, evidencebased guidelines are lacking.
3. Economics—likely, the cheapest way to make the histologic diagnosis is to try and make the diagnosis on an H&E stain when there may be no inflammation or organisms and others with a typical Helicobacter pattern of inflammation and readily identifiable organisms. However, if inflammation is present but organisms are not identified, it is best to do a special stain. The choice of the stain is a matter of personal choice, available resources, and emphasis on TAT. One could start with a “quick and cheap” stain (e.g., Giemsa stain) that, if positive, solves the problem, but if it is negative or equivocal, one can order a more sensitive and specific stain (silver stain or immunostain) or alternatively do the more expensive but sensitive stains at the outset. Unfortunately, in this regard, any data on cost-efficiency are lacking.
4. Clinical factors—if the patient has a bleeding ulcer, this is far more critical than when biopsies are taken for long-standing symptoms, and the patient is returning in 2 weeks for the results.
Special stains forH. pyloriidentification— which is best? Most pathologists use H&E plus a second stain for H. pylori visualization. The appearance of the organism varies with the staining technique used. Commonly used special stains can be divided arbitrarily into (a) silver-based stains, (b) non-silverbased stains, and (c) immunohistochemistry. Silver stains are more expensive, but organisms appear larger and there is contrast with the background, so small numbers of organisms are easier to identify. With few exceptions, non-silver-based methods try to detect small blue organisms on a blue background, so unless organisms are numerous they can be difficult to detect. Immunohistochemistry is an alternative to silver stains, primarily because the organisms are readily identifiable. It has the potential advantage of not picking up other organisms that may be present in the stomach (enteric, oral), which can proliferate in patients with atrophy or who are taking PPIs. Some labs use an immunostain initially, while others use a silver stain. Pathologists are faced with several types of stains to choose from. No stain is perfect.
Table 13-6 Stains for Detection ofHelicobacter pylori
Sometimes high background (deposits); epithelial and lymphoid nuclei can look atypical; H&E necessary
Warthin-Starry (or modified Steiner)
High
28 min
High
Sometimes high background (deposits)—autostainers tend to be much cleaner and reliable; epithelial and lymphoid nuclei can look atypical; H&E necessary
Stains are listed in alphabetic order. Other stains used for H. pylori identification but not included in this table include Half-Gram, Brown-Hopps, Toluidine Orange, Acridine Orange-ultraviolet fluorescence, and Butler Modified Wright stain.
Pathology laboratories vary in their resources. Silver-based stains and immunohistochemistry stains can be ideal in some laboratories but a technician’s (and a pathologist’s) nightmare in others. The best stain is the one that works in your laboratory bearing in mind that an H&E stain interpreted by inexperienced pathology staff is the least sensitive and the least specific, although frequently possible, while absolutely normal biopsies can similarly be identified that do not require a special stain. Silver and immunostains done well are the most sensitive and specific. A special stain is recommended to facilitate H. pylori diagnosis (Table 13-6). However, the best stain in the world is of no value if the biopsies provided are insufficient or inadequate to answer the question “Does the patient have Helicobacter gastritis?”
(a) Silver-based stains produce dense, black, fine deposit of silver and silver oxide where the silver ions have been reduced. Silver impregnation makes H. pylori appear larger, making detection easier (Fig. 13-25A-C). This is particularly useful in patients on PPI where bacteria present are fewer in number and generally smaller in size. The Warthin-Starry and the modified Steiner stains are commonly used. In addition, silver techniques are so sensitive that they can sometimes give nonspecific background deposits (“dirty preparations”). Background deposits are less frequently seen with El-Zimaity dual stain169 and the Genta stain,170 or on some automated staining machines. The El-Zimaity dual stain was introduced for the simultaneous visualization of the bacteria and gastric metaplasia in the duodenum. However, since gastric mucin is always positive for periodic acid-Schiff (PAS) (glycoprotein), this stain has proven useful when only few bacteria are present.171 The Genta stain is particularly useful in high-workload laboratories as only one slide is reviewed (instead of H&E and another stain); the stain can be technically challenging in some laboratories.172 For laboratories that cannot use uranyl nitrate, the modified Genta stain173 can be used instead; this uses lead nitrate as a substitute, together with Alcian blue and an H&E stain. In general, background deposits are less frequently seen with slides processed using the autostainer. Silver-based methods have the disadvantage of being relatively more expensive, cannot be discarded into the drains as it is a heavy metal poison, and the stain does not work in improperly fixed tissue (e.g., if the formalin used is too diluted).
Figure 13-25.Helicobacter pylori as demonstrated by a variety of stains. The first three are silver stains—A: Genta. B: El-Zimaity Dual PAS Silver. C: Warthin-Starry. The second three are nonsilver stains—D: Diff-Quik.
Figure 13-25.(Continued)E: Alcian yellow. F: Triple stain with carbol-fuchsin.
(b) In non-silver-based stains, (Fig. 13-25D-F), the dye tints the bacteria with a color. For example, the bacteria are blue with Diff-Quik, crystal violet, and Giemsa stains.168, 174 In contrast to silver-based stains, visualizing the bacteria totally depends on the contrast between the bacteria and tissue sections. The Diff-Quik has the better sensitivity and specificity; the Giemsa stain has suboptimal accuracy (more false-positive results)168, 175 especially with less experienced pathologists. The lack of contrast between organism and background renders specimen evaluation for H. pylori infection rather arduous, which is reflected in the time required to examine slides, especially when multiple biopsies are present and several serial sections on the slide may need to be examined to be convinced that H. pylori is either present or absent.168
In this group, we prefer Leung’s Alcian yellow stain;176 it is easier to spot blue (but still small) bacteria against yellow gastric mucin, compared with blue organisms in paler blue mucin with other stains. These stains are relatively inexpensive and thus are commonly used. Though we prefer silver-based stains, tinctorial stains do not require optimum formalin fixation. This group of stains is particularly disadvantaged by mucus and debris that can mimic the presence of H. pylori. In equivocal situations (common with chronic PPI use), pathologists may err on the side of overcalling the presence of any bacteria present as H. pylori. This might seem unimportant. However, H. pylori infection is absent in up to 27% of patients with endoscopically proven duodenal ulcers.177 Such patients appear to have a significantly worse outcome when treated empirically for the infection,178 almost certainly because they were also ingesting NSAIDs.
(c) Immunohistochemistry is perceived as the gold standard by some investigators as in some settings immunohistochemistry reduces the false-positive rate, providing greater accuracy over routine histochemistry.179 Both polyclonal and monoclonal antibodies are commercially available. In addition, in some laboratories, immunohistochemical stains are performed using an autostainer, which translates into less technical time and less technical training. It is our experience that cases with few bacteria can also be missed with immunostains. We have also seen other Helicobacter species (H. heilmannii) stain positive with antibodies (especially polyclonal) for H. pylori, and morphologic differences in their spiral morphology are difficult to detect on immunostains. Importantly, anti-H. pylori antibodies also stain the coccoid forms of the bacteria. These forms may possibly be viable but are nonculturable, and one study suggests less virulent, and less likely to colonize and induce inflammation.180 Coccoid forms have never (yet) been shown to be viable; further, coccoid organisms can come from the small bowel and oral cavity, so basing a diagnosis of Helicobacter gastritis only on coccoid forms of organisms should arguably never be made, or at least should be stated clearly in the pathology report. Similar to silverbased methods, optimal specimen processing (i.e., formalin fixative) is necessary for immunohistochemistry. Fluorescent antibodies are unpopular because in addition to requiring the use of fluorescent microscopes, they fade over time, and so do not provide a permanent record.
Role of Biopsy Site(s) in Determining H. pylori Infection While histology may be considered a gold standard, the reliability of detecting H. pylori infection is actually dependent on the site, number, and size of gastric biopsies, as well as stain used and expertise in staining to visualize the bacteria. Falsenegative and -positive interpretations are all too easy. The standard of two biopsies from the antrum and two from the oxyntic mucosa covers most eventualities. Stripping of surface epithelium, that seems to occur readily in oxyntic mucosa, is a good reason to always take 2 oxyntic biopsies. Some use a combination of three biopsies (angulus, greater curvature of the corpus, and greater curvature of the antrum) (see Fig. 13-2), which seems to be the minimum for accurate diagnosis.136 When biopsy specimens had few H. pylori per slide, the same patient had biopsies from other areas with more bacteria. This emphasizes the need to obtain multiple biopsies to exclude H. pylori infection.136 In patients treated with PPIs, antibiotics or other antibacterials such as bismuth salts 2 to 4 weeks prior to biopsy, the bacteria may be restricted to the corpus or fundus, and as small numbers of organisms may be present, really need a silver or immunostain if not detected using other stains.
Failure to Biopsy Both Oxyntic and Antral Mucosa— Major Cause of Failure to Detect Helicobacter When biopsies are taken for Helicobacter detection, it becomes important to ensure that both antral and oxyntic mucosa are biopsied, as when the organisms are present, biopsying only one site is a major reason for failure of detection. It will be recalled that H. pylori infection is initially primarily an antral disease, and oxyntic mucosa may show only minimal superficial inflammation, and organisms may be impossible to detect. Conversely with time, atrophy of oxyntic mucosa, or PPI therapy, may result in the organism moving proximally, at which point it may no longer be detectable in the antrum. When only antral or oxyntic mucosa is biopsied, the false-negative reading for Helicobacter is about 10% to 15% (El-Zimaity H e-pub Canadian J Gastroenterol).
Major reasons for failing to obtain both antral and oxyntic biopsies when the endoscopist believes both sites have been biopsied include the following:
1. The endoscopists believe that the antrum is the preferred site for Helicobacter, so never biopsy the oxyntic mucosa.
2. Many endoscopists do not appreciate that the histologic antrum is often quite distal in the anatomical antrum, so that the prepyloric region needs to be biopsied to guarantee getting antral mucosa, although this may also fail if the anrum has diffuse intestinal metaplasia. Some endoscopists always attempt to obtain biopsies from both antral and oxyntic mucosa, but others tend to obtain three oxyntic and one antral or all oxyntic mucosa, and are quite surprised when the “comment” states that all biopsies are from one site— invariably oxyntic when “random gastric biopsies” are taken.
3. If there is an atrophic front that passes unrecognized endoscopically (Fig. 13-21), and the mucosa is also not recognized as being atrophic, and it is as biopsied a presumed oxyntic mucosa, then the latter will not be obtained.
Differential diagnosis of the H. pylori organisms. Chains of cocci may be seen and only occasionally are confused with H. pylori. It is possible that in some instances these cocci represent degenerate forms of the organisms.180 Confusion with enterococcal gastritis, which is rare, can occur, primarily if one is unaware of its existence, and when immunostains are not used67 (see Fig. 13-15). Numerous organisms, often mixed cocci, rods, and even fungi can be seen on patients on long-term PPIs (Fig. 13-26). Determining whether Helicobacter are present amongst an admixture of organisms is one situation in which an immunostain is far superior to all other blanket bacterial stains including silver stains.
Another artifact is that the apices of mucous cells may be cut in a plane such that they may look like a curved organism, but always in the same direction, so that only one-half of the “wave” is present. Helicobacter heilmannii are larger (2.5-10 µm) than H. pylori and are much less frequently observed. These organisms contain four to nine even spirals along their length and are frequently arranged in stacks.
Figure 13-26. Organisms that can be found in patients on PPIs that must not be misinterpreted as H. pylori. They may come from swallowed oral organisms or small bowel organisms refluxed into the stomach. A: A plethora of organism including fungi seen on an H&E stain. B: Giemsa stain with myriad organisms in the lumen. C: Warthin-Starry stain. The two panels on the left (C1 and C2) have single organisms that could be considered to be Helicobacter. In the third panel (C3) these coccoid organisms could be misinterpreted as coccal forms of H. pylori, but in the absence of morphologically typical forms that interpretation should not be made. D: Filamentous and bacillary organisms visible on H&E stain.
Algorithm for Failing to Detect Helicobacter When the Morphology Suggests That They Should Be Present It is not infrequent to have gastric biopsies with a clinical question to rule out H. pylori (“R/O Hp”), and be faced with a morphology that suggests that Helicobacter should be present, but special stains fail to detect the organism. It is useful to have an algorithm for dealing with such biopsies. All of these are discussed in various sections of this chapter, but have been compiled together here for convenience.
1.Was an appropriate special stain ordered? (just H&E is unreliable.)
2.False-negative stain. If a blue-on-blue stain (Cresyl violet, Giemsa, Diff-Quick, etc.) was carried out, do a silver, dual, or immunostain.
3.PPI changes. If oxyntic mucosa is present, are there changes of PPI ingestion? PPIs can reduce Helicobacter to undetectable levels. Sometimes they may only be present deep in the oxyntic mucosa, sometimes only within parietal cells, when either silver or immunostains are usually necessary to detect them (see Fig. 13-22). Recent administration of PPIs however rapidly reduces numbers of Helicobacter, but the morphologic changes are not detectable on histology as it takes weeks for these changes to develop, so requires clinical input. Because PPIs may result in numerous organisms being able to grow in the stomach—both oral and duodenal, a Helicobacter immunostain is the stain of choice for determining if Helicobacter are present in and admixture of bacteria.
4.Lymphocytic gastritis. In addition to the lamina propria infiltrate, and especially if this is active (neutrophilic), is there an intraepithelial lymphocytosis? If so, lymphocytic gastritis frequently has no Helicobacter, but serology is positive, and treatment with eradication therapy results in healing of the gastritis. This form of lymphocytic gastritis is usually easily distinguished from the mild intraepithelial lymphocytosis associated with celiac disease.
5.Diffuse reactive gastropathy. Organisms are rarely found in the absence of mucin, so when there is diffuse reactive (chemical) gastropathy, Helicobacter are rarely found. In some conditions such as post-gastric resection or gastroenterostomy, there is pathologic duodenogastric reflux with resulting diffuse gastropathy. Even if Helicobacter are present proximally, marked mucin depletion can prevent their identification in the part of the stomach affected by reflux.
6.Sampling. Are both antral and oxyntic mucosa present? If not, this could well be the reason as 10% to 15% of patients with Helicobacter have them in only one site (El-Zimaity, E-pub Canadian J Gastroenterology).
7.Intestinal metaplasia. Helicobacter will not grow in intestinal type mucosa whether native or metaplastic. So if, for example, two antral biopsies are taken and both are completely metaplastic, no organisms may be detected. Unless the proximal mucosa is also sampled, the organisms will not be found.
8.Atrophy with pseudopyloric metaplasia imitating antral mucosa without (or with minimal) intestinal metaplasia. Helicobacter are not found in either the residual antral or metaplastic mucosa. However, to recognize this requires the ability to distinguish antral mucosa from oxyntic mucosa that has undergone complete atrophy with pseudopyloric metaplasia. This is usually part of the creeping atrophy that takes place in Helicobacter gastritis with time, rather than autoimmune gastritis.
If all biopsies appear to be antral (nonoxyntic), a hint to help identify pseudopyloric metaplasia is that one needs to evaluate if all biopsies are similar regarding inflammation and architecture. If different (e.g., one/some are inflamed or architecturally different—especially holes between the glands [atrophy] and one/some are noninflamed or with regular architecture), the likelihood is that the inflamed biopsies, which invariably have a degree of gland loss, represent oxyntic mucosa with atrophy and pseudopyloric metaplasia. An immunostain for gastrin will show the antral biopsies by the presence of G cells, while their lack indicates atrophic oxyntic mucosa. If gastrin stain is not available, then chromogranin A stain can serve a similar function, but now the distribution of endocrine cells needs to be evaluated. G cells primarily form a band below the mucous neck, with few or no cells in the pyloric glands. Conversely oxyntic mucosa has regularly spaced endocrine cells if normal, but with long-standing hypergasrtinemia resulting in their hyperplasia, these form chains (linear hyperplasia) and small clusters of endocrine cells (microcarcinoids), and they also migrate to the base of the crypt close to the muscularis mucosae. In some patients, these changes are detectable on H&E stain as the crypt bases can develop a double layer of nuclei, the outer layer being the endocrine cells.
9.Other causes of diffuse gastritis
(a) Infections—CMV, syphilis, and other bacteria (Micrococcus). Immunostains to CMV if not visible on the H&E sections and Treponema pallidum (together with good clinical suspicion) are required for these to be considered.
(b) An up-regulated immune system—IBD, especially Crohn’s disease, in which a modest diffuse chronic superficial inflammation is often present. Appropriate clinical input is required for this.
10.Recent administration of antibiotics or Helicobacter eradication therapy. (This clearly needs clinical input.) The chronic inflammation following Helicobacter eradication can take months and sometimes over a year to completely subside. It is likely that rare instances of spontaneous eradication occur, or that eradication occurs when patients are put on antibiotics or other reasons. Either way, the chronic inflammation persists for a time.
11.Idiopathic gastritis. If the above have been excluded, there remains a group of patients in whom there appears to be no known cause despite extensive investigation. Pathologists seeing gastric biopsies regularly are well aware of this subgroup as it is not small and may account for perhaps 15% to 25% of biopsies. It therefore has to be acknowledged that there are a group of patients with chronic (less commonly chronic active) gastritis with no known cause. Fortunately, most of the gastritis is relatively mild and is Helicobacter negative in multiple biopsies and serologically. These are signed out simply as “chronic gastritis; Helicobacter are not identified”. However, this diagnosis should not be used casually and until all other identifiable causes of inflammation resembling Helicobacter have been excluded as outlined above.
Figure 13-27. Atrophic antral gastritis. A: Atrophic antral gland gastritis. Much of the mucosa has been replaced (left two-thirds) by intestinal metaplasia and inflammatory cells in the lamina propria. Goblet cells stain blue in this H&E-Alcian blue, pH 2.5 stain. There are only a few residual antral mucous glands at the extreme right of the picture. B: Villiform normal antral gland mucosa (uncommon but not rare) should not be confused with intestinal metaplasia. The surface and pit epithelial cells are of the normal gastric mucous type. G-cells in the mucous neck region are not visible at this magnification. C: Complete atrophic gastritis of the antral gland mucosa with intestinal metaplasia occupying the full thickness of the mucosa.
Atrophic gastritis and gastric atrophy.Atrophic gastritis refers to the finding of variable gland loss resulting from mucosal inflammation, often associated with metaplasia to either pyloric or intestinal types of mucosa. Although this term is most frequently applied to oxyntic mucosa, it can be applied to oxyntic mucosa, antrum, or both. In the antrum, atrophy is often associated with intestinal metaplasia that occupies the full thickness of the mucosa in all or part of a biopsy (Fig. 13-27). Complete or almost complete replacement of the antrum with intestinal metaplasia is associated with a higher cancer risk, even in the absence of corpus atrophy.150
Corpus atrophy begins at the junction between fundic and the antral mucosa.85, 181, 182 This usually takes the form of loss of oxyntic glands with pseudopyloric, intestinal, and sometimes pancreatic metaplasia.85, 150, 182 Corpus atrophy shifts proximally from the vicinity of the angulus circumferentially, such that the antrum appears to expand with advancing atrophic gastritis.181, 183 Corpus atrophy progresses at about the same rate in the contiguous greater curve, proximal half of the lesser curve, and neighboring anterior and posterior walls of the corpus.150, 181
The rate of progression of H. pylori gastritis depends on the acid milieu. The natural history of H. pylori gastritis is to go through a cascade of events from nonatrophic gastritis, degrees of atrophic gastritis, and in some patients, dysplasia.184, 185, 186 The atrophy involves loss of oxyntic glands with extension of mucous neck cells down into the pits (mucous cell hyperplasia—historically considered to be pseudopyloric metaplasia but the name persists). Intestinal metaplasia usually supervenes within this epithelium. However, on occasion isolated intestinal metaplasia may happen without adjacent pseudopyloric metaplasia. Presumably this is the result of focal injury, possibly related to NSAIDs or ASA, etc.
To increase our likelihood of identifying corpus atrophy—when present—it is important to remember five principles:
1. Corpus atrophy begins at the antrum-corpus junction. In the early stages of atrophic gastritis, for example, in children,85 the location of early atrophy is only just proximal to the normal histologic antral-corpus border so that unless biopsies are taken from this region the atrophy will be missed (Fig. 13-22).120
2. Atrophy is gland loss with or without its replacement with fibrosis or metaplastic epithelium.150, 181, 187
3. The atrophic border extends proximally at a similar rate on both curvatures. However, because the lesser curve is much shorter than the greater curve, locations high on the greater curvature are among the last to undergo atrophy150, 181, 187, 188; it is therefore also the best site to biopsy to find residual Helicobacter infection.
4. The presence of a dense mucosal mononuclear cells deep in the lamina propria is a predictor for the presence of gastric atrophy as the acid level will already be low.189, 190
5. As the amount of atrophy increases, acid secretion diminishes. This stimulates G cells to increase gastrin secretion, G cells become hyperplastic, and there is hypertrophy of remaining parietal cells proximally, but a variable increase in the number of ECL cells in the body mucosa. This may be undetectable in residual oxyntic mucosa, but in the atrophic mucosa, there is ECL hyperplasia that varies from indiscernible to hyperplasia. It is basal and immediately above the muscularis mucosae, then linear hyperplasia; microcarcinoids may then form. However, while the tell-tale double row of nuclei may be visible above the muscularis mucosae, endocrine stains, especially chromogranin A (Fig. 13-28), and lack of gastrin staining, if that is available, help to make the diagnosis.
Recognizing Pseudopyloric Metaplasia (Mucous Cell Hyperplasia) The normal oxyntic mucosa has straight glands composed of tightly packed chief and parietal cells, endocrine cells, and mucus cells. There is a higher ratio of glands to foveola (4-5:1) than the antrum (1-2:1). With continuous inflammation, there is a progressive loss of specialized cells. Eventually, the oxyntic glands are replaced by mucous cells extending down from the mucous neck regions, resembling antral/pyloric glands (pseudopyloric metaplasia), and failing to differentiate into parietal, chief, and endocrine cells (Fig. 13-28). The diagnosis of pseudopyloric metaplasia can be a diagnostic challenge as it is readily interpreted as antral mucosa. The diagnosis of pseudopyloric metaplasia, and its distinction from antral mucosa, can be facilitated by several methods (Fig. 13-28).
1. Because G cells are never seen in the body,191 (other than rare cells in intestinal metaplasia), the absence of G cells is particularly useful in recognizing pseudopyloric metaplasia, or conversely the presence of G cells indicates that the mucosa is antral (Fig. 13-29). Gastrin immunostains are the easiest way of doing this.
2. Pepsinogen I (PGI), if available, is localized in chief cells, mucous-neck cells, and transitional mucous neck/chief cells of the human oxyntic mucosa192; it is not localized in antral gland cells (Fig. 13-29). Thus, it can also be used to differentiate antrum from oxyntic mucosa.191
3. Use both—ideal. Pseudopyloric metaplasia is identified by the presence of mucosa that superficially resembles antrum, stains positive for PGI, stains negative for G-cells, and is anatomically in a region where corpus would be expected85, 150 (Fig. 13-29).
4. The distribution of endocrine cells that can be highlighted with endocrine markers (chromogranin or synaptophysin—whichever works best) in the antrum and atrophic corpus mucosa can also help in the differentiation (Fig. 13-30). The normal endocrine cell staining in the antrum is intense, almost linear beneath the mucous neck region but above the bases of the crypts, while in oxyntic mucosa with pyloric metaplasia, it is initially represented by single cells diffusely scattered throughout the glands, but with progression there is increasing hyperplasia at the bases of the glands.
Figure 13-28. Two biopsies from the same patient to “rule out Hp.” That on the left (A) is normal antral mucosa, and that on the right (B), which is clearly not oxyntic, looks like inflamed antral mucosa. Note the architectural disarray in B at low power compared to A, as well as the additional inflammation. C: Numerous readily visible endocrine cells (mainly G cells—arrows) confirm that this is antrum. By contrast in (D), the endocrine cells form a double layer (at least in this patient) indicative of ECL-cell hyperplasia at the bases of the crypts immediately above the muscularis mucosae. These appearances are confirmed in both biopsies using chromogranin. In (E), there is the usual band of endocrine cells seen in the antrum in the mid-third of the mucosa with rare deeper cells (a gastrin stain would have been even more specific for antral mucosa). In contrast in (F), the endocrine cells are at the base of the mucosa, and here they form chains and completely encircle some pits, indicative of quite marked ECL hyperplasia. The gastrin stain was completely negative.
Figure 13-29. Normal corpus (oxyntic) mucosa in the top three panels and antral mucosa in the lower three. PGI is produced only in chief cells of the oxyntic mucosa and gastrin only in the G-cells of the antral mucosa. In practice G-cells can be recognized in H&E sections because of the perinuclear halos just below the mucous neck region.
Pseudopyloric metaplasia (also called pyloric metaplasia or mucus metaplasia and ulcer-associated cell lineage13, 85, 150, 193 was described as early as 1959188 in benign gastric ulcers proximal to the normal border zone (antrum-corpus junction). In fact, prior to the rediscovery of H. pylori, a proximally advancing atrophic front with pseudopyloric metaplasia was considered part of the normal gastric aging process.181 Following the rediscovery of H. pylori, an association was demonstrated between the presence of mucous glands in corpus biopsies and the age of H. pylori-infected patients. Pseudopyloric metaplasia (downward extension of mucous neck cells) is considered regenerative in nature194, 195 and is also observed in gastric remnants following distal gastrectomy with gastroenteric anastomosis forming a “neo-antrum” although without new G-cells assuming that the antrum was completely removed.196
Figure 13-30. Use of standard endocrine stains to identify site by pattern of immunoreactivity. In normal oxyntic mucosa (top 2 panels) endocrine cells are distributed fairly regularly and singly. In antral mucosa G-cells form a zone in the midpart of the mucosa (lower right). Further, in atrophy with severe pseudopyloric metaplasia the atrophy results in hypergastrinemia and subsequent G-cell hyperplasia that affects the pits immediately beneath the mucous neck zone (bottom left).
In interpreting gastric biopsies, recognizing biopsy location and the location of the transitional zone is paramount. Although finding transitional mucosa (where antral mucosa becomes oxyntic which can be gradual or abrupt—also known as junctinal or intermediate mucosa) can help in determining the location of early atrophy, its recognition is not always obvious, especially on H&E sections. As discussed above, G cells are never seen in the body.191 The complete disappearance of G cells is probably the most reliable method to define the proximal border of the antral-body transitional zone. Other features including the change from single tubular glands in the body to branched glands in the antrum. The disappearance of parietal cells is not reliable as parietal cells can be normally present in the antrum and may extend to the duodenal junction.
Making the Diagnosis of Atrophy of Oxyntic Mucosa, Especially without Intestinal Metaplasia The recognition of atrophic oxyntic mucosa with pseudopyloric metaplasia requires suspicion, experience, and the ability to confirm the diagnosis using immunohistochemistry.
Recognition. Recognizing pseudopyloric metaplasia should initially be endoscopic, for the atrophic front (see Figs. 13-21 and 13-22) should be recognized by this modality, so it may inadvertently be biopsied as oxyntic mucosa. Histologically it requires the ability to distinguish atrophic oxyntic mucosa from antral mucosa (Fig. 13-28). The three most useful features are the presence in atrophic oxyntic mucosa of (a) disordered pseudopyloric glands, (b) chronic inflammation, and (c) endocrine cell changes. The initial clue is to routinely look for G cells in H&E sections, where they are readily visible in antral mucosa beneath the mucous neck region (Figs. 13-28 and 13-29). With pseudopyloric metaplasia, G cells are absent. Endocrine cells in oxyntic mucosa are much more subtle to find on H&E sections. Native ECL cells are virtually invisible and initially retain the fairly diffuse and uniform pattern of endocrine cells seen in normal oxyntic mucosa on immunostaining, As expected, immunostains for gastrin are negative. With increasing serum gastrin levels, there is hyperplasia of endocrine cells that move to the pit base. This progresses to linear hyperplasia, and when this is marked, it can be recognized on the H&E stain by a double layer of nuclei, the outermost layer being pure endocrine cells, along with some of the inner layer (Fig. 13-28). Endocrine micronests often follow or are already present.
Conversely, antral mucosa is usually uniform in the distribution of antral glands; its G cells are readily recognized on H&E sections and are frequently hyperplastic, similar to the changes seen in patients on long-term PPIs as the mechanism (prolonged hypochlorhydria) is identical (Figs. 13-28 and 13-29). In the presence of atrophic oxyntic mucosa, there is usually little or no chronic inflammation in the antrum, and if present is usually considerably less than that in the atrophic oxyntic mucosa, as all of the Helicobacter-related inflammation has moved far proximally, and the pattern of endocrine cells is completely different. G cells can invariably be recognized and most of the time are overtly hyperplastic. This can be demonstrated by gastrin stains or the distribution of endocrine cells primarily in a band in or just above the mid-part of the mucosa. Thus, both types of mucosa can be recognized on H&E sections, although it takes most considerable time to understand this and remember to look for these changes in routine biopsies. When the antrum is not only atrophic but has diffuse intestinal metaplasia, defining antrum can be impossible unless residual clusters of G-cells can be identified.
When biopsies from both antral and corpus types of mucosa are present together, usually to “rule out H. pylori,” the clue is that while initially all biopsies look like antral mucosa, it is usually apparent that there are two different biopsy types (Fig. 13-28). One has the features of uninflamed or minimally inflamed antral mucosa with appropriate G-cell populations, the other more inflamed with patchy pseudopyloric glands, no G-cell band, and possibly endocrine cell hyperplasia at the pit bases. There is virtually no recognized disorder that produces such variability in antral biopsies, so this combination of changes should immediately raise a red flag that at least one of the biopsies is not antral in origin.
Numerous variations in biopsy combinations can be found but the most common variants are the following: (a) the antral mucosa may be as inflamed as the atrophic mucosa, but all of the other features are present; (b) occasionally one of the oxyntic mucosal biopsies is proximal to the “atrophic front,” so the biopsies consist of antral, atrophic oxyntic mucosa and either normal, but more commonly inflamed oxyntic mucosa, often with Helicobacter organisms; (c) occasionally the antral biopsies may not be present at all, so the biopsies are either all atrophic oxyntic alone or atrophic oxyntic and oxyntic. In our experience, this is uncommon.
Distinction from autoimmune gastritis. As discussed subsequently, the diagnosis of autoimmune gastritis (AIG) is often best made by a combination of the changes just described, together with serology, with antibodies to intrinsic factor and parietal cells, the latter also being present quite frequently in Helicobacter gastritis. However, it may also be appreciated because of the extent of disease (see subsequent section on autoimmune gastritis and Fig. 13-57). Although one might predict that AIG would occur diffusely throughout the oxyntic mucosa bearing the stomach, like other autoimmune conditions there is focality, at least until there is complete metaplasia. As it appears that AIG frequently results from Helicobacter, if Helicobacter have not been identified, serology for the organism is also appropriate as it is possible that its eradication may prevent further progression of the disease.
Intestinal Metaplasia Intestinal metaplasia comes in two major forms that often coexist, complete and incomplete (Fig. 13-31). The complete form essentially resembles the colon, with absorptive, goblet, Paneth, and endocrine cells, but when well developed may resemble small intestine with villi. Incomplete intestinal metaplasia consists of gastric mucous-producing cells with goblet cells interspersed amongst them. Intestinal metaplasia is a required precursor for intestinal-type carcinoma. The development of gastric carcinoma is a slow and unpredictable process, and intestinal metaplasia is an easily recognizable marker for atrophy.
Subtyping intestinal metaplasia using high iron diamine staining was thought to identify subgroups of patients with different risk potential (Fig. 13-31). Intestinal metaplasia subtype III (black-staining sulfomucin in goblet cells of incomplete intestinal metaplasia) is often considered the highest-risk precursor lesion for the intestinal form of gastric cancer,197, 198, 199, 200 but this is not uniformly accepted.
In addition, approximately equal number of studies have suggested that intestinal metaplasia regresses or does not regress after H. pylori treatment.182, 201, 202, 203, 204, 205, 206, 207, 208 Sampling error is the likely factor responsible for this discrepancy. This question is a critical one, the corollary being whether eradication of Helicobacter prevents progression of intestinal metaplasia, but most importantly the associated cancer risk. Is there a point of no return? The assumption is that the greater the extent of metaplasia the greater the cancer risk, even though virtually all of the carcinomas arising from this etiologic pathway are in the antrum and lesser curve. If Helicobacter eradication prevents the risk, then all of the grading systems for atrophy are obsolete. However, currently this is still unclear, so a grading system for atrophy still has to be considered (see subsequent discussion).
Figure 13-31. Types of intestinal metaplasia. Complete type refers to metaplastic glands composed of goblet cells, columnar cells with a distinctive brush border and in some cases Paneth cells. The type 1 goblet cell of the small intestine epithelium is the predominant element and is located chiefly in the villi (A), while the type II goblet cell is preferentially located in the crypts. Using HID-AB stain, goblet cells are blue in type III whereas columnar cell mucus is blue. In type IV both the goblet cells and columnar cell mucus are sulphated (brown-stainnig).
Prior studies suggesting an association of type III intestinal metaplasia with the development of gastric cancer197, 198, 199, 200 did not take into account the higher prevalence of incomplete intestinal metaplasia (type III) in the gastric antrum.23, 150, 197, 198, 199, 200, 209 In practice, areas of intestinal metaplasia (or a certain subtype) are generally small and can easily be missed at follow-up.150, 182
A small percentage of cancer patients show complete replacement of the antral mucosa with intestinal metaplasia and have normal-appearing oxyntic mucosa.150 It is unknown if these individuals lose their G-cells and have normal or reduced acid secretion. Continued inflammation with antral atrophy could possibly lead to sufficient destruction of G-cells,210 which can result in a fall in acid secretion.211, 212 Alternatively, contiguous sheets of intestinal metaplasia may be an unstable epithelium especially upon exposure to persistent low-dose dietary or salivary carcinogens, and increase the risk of dysplasia and carcinoma.
Overall, it is apparent that it is not possible to make recommendations or prognoses-based comments on biopsies (single or multiple) showing sulfomucin expression in areas with intestinal metaplasia.182, 213, 214, 215 All data suggest that the extent of mucosal atrophy within a region of the stomach is more important than the type of intestinal metaplasia in the development of intestinal type of gastric cancer.
While intestinal metaplasia is a form of atrophy that is easy for pathologists to recognize, it is also important to determine whether intestinal metaplasia is present as an isolated patch within nonatrophic mucosa or amidst an atrophic lawn.120, 150 Thus a patch of intestinal metaplasia in nonatrophic mucosa is a reparative phenomenon, and there are no data to suggest that it is associated with an increased risk of carcinoma.
Changes Associated with Long-term Proton Pump Inhibitors and Corpus Atrophy PPIs are potent inhibitors of gastric acid secretion. They are widely used in the treatment of acid-peptic diseases and effectively alleviate acid-peptic symptoms and facilitate healing of inflamed or ulcerated mucosa. These drugs are increasingly used long term, frequently for a lifetime, in patients with typical or atypical symptoms of GERD, and in NSAID or aspirin users, such as patients with rheumatoid or osteoarthritis.216 They give rise to the typically hypertrophied PCs that result from hypertrophy of endoplasmic reticulum (Fig. 13-32). This change is readily visible at scanning power because of the serrated, rather than round, shape of the pits lumen.
Although 20% to 50% of patients on long-term PPIs may still be infected with H. pylori, eradication of Helicobacter is not always carried out prior to starting PPIs. In these patients, Helicobacter pylori accelerates corpus gastritis with subsequent low acid secretion resulting from both the PPIs and the ensuing atrophy and resulting low acid secretion.151, 152, 153, 217, 218, 219, 220, 221, 222, 223, 224, 225 This in turn facilitates proximal migration and increased severity of H. pylori gastritis.151, 153, 189, 226 It also allows the development of a corpus-predominant gastritis rather than a pangastritis.227 PPI therapy is associated with a reduction in bacterial load, both in the antrum and in the corpus, and a tendency for antral histology to improve and corpus gastritis to either not change or worsen. With PPI therapy, there is a significant progression of the inflammatory reaction deeper within the pit to involve the proliferative zone189 (see Fig. 13-23) as seen in atrophic gastritis. This may be related to the migration of the bacteria deeper into the oxyntic glands secondary to PPI therapy.
Figure 13-32. Hypertrophy of parietal cells as seen in longstanding hypergastrinemia. By far the most common cause of this is long-term ingestion of PPIs.
Prolonged PPI use in H. pylori-negative patients showed no difference in gastric inflammation or atrophy over a 7-year period.228 In contrast, H. pylori-positive patients followed over the same period showed increased inflammation and gastric atrophy.228 Gastric atrophy with peripheral neuropathy and vitamin B12 deficiency has been described following 20-year treatment with PPI without H. pylori eradication.229 These studies highlight the importance of considering H. pylori eradication in infected, long-term PPI users.
As PPI therapy reduces gastric acidity, it also reduces H. pylori density (average number of H. pylori per biopsy) in the stomach, H. pylori may not be identified in gastric biopsies.219, 230, 231 One has to rely on the diffuse pattern of inflammation to diagnose the infection. Further, the Helicobacter seek out the acid to the point that they may invade the pits, and can even be found in the canaliculi of PCs (Fig. 13-33). Histopathologic features suggestive of long-term PPI use include not only PC hypertrophy but a degree of enterochromaffin-like (ECL) cell hyperplasia from long-standing hypergastrinemia, although this requires endocrine stains, and often counting to demonstrate the changes. These changes therefore resemble changes seen in the stomach in patients with Zollinger-Ellison syndrome, even to the point of small ECL-tumors.
Figure 13-33.Helicobacter present deep in the pits and also in the canaliculi of PCs, some of which are arrowed. (Courtesy of Dr. M Vieth.)
PPI therapy, particularly with long-term or highdose administration or both, is associated with several potential adverse effects, including vitamin B12 deficiency irrespective of H. pylori status,229, 232, 233, 234 enteric infections (e.g., C. difficile), community-acquired pneumonia, osteopenia and hip fractures, and small intestinal bacterial overgrowth. Proposed mechanisms are that reduced acidity impairs cobalamin release from dietary protein and bacterial overgrowth increases competitive consumption.229, 233, 234
Overgrowth of Other Organisms in the Hypo or Achlorhydric Stomach It is not uncommon to see a variety of cocci and bacilli in gastric mucosa of patients on high-dose long-term PPIs (see Fig. 13-26) or with extensive atrophy from prolonged H. pylori. These organisms are either of oral or duodenal origin as PPIs can increase the median gastric pH from about 3.25 to 6.75 (Fig. 13-34). These should not be mistaken for H. pylori or its coccoid forms. While these colonies can be associated with inflammation, it is often difficult to be sure that the patient does not have Helicobacter that is being suppressed by the PPIs. Helicobacter immunohistochemistry is the obvious way of resolving this issue, but the typical morphology of Helicobacter needs to be present to make the diagnosis.
Staging gastric atrophy. Gastric atrophy and atrophic gastritis are often used synonymously in practice, although atrophic gastritis is the process resulting in atrophy. It also represents transmucosal inflammation (as opposed to superficial gastritis) that is likely to progress to glandular atrophy. Gastric atrophy simply refers to diffuse loss of gastric glands. As the presence of gastric atrophy, with or without intestinal metaplasia, is the critical determinant of a person’s risk for gastric cancer,120, 235, 236 recognizing its degree does have clinical implications. Most intestinal and some diffuse types of gastric carcinomas arise on a background of atrophic gastritis; an index of the extent of atrophy can be useful in predicting patients at greatest risk for carcinoma.118, 184, 237 Screening protocols include endoscopic screening, serum biomarker tests, and histology.
Figure 13-34. Principal organisms cultured from the stomach in patients taking omeprazole. Note the numbers of coccoid or cocco-bacillary organisms included. It is therefore important that these not be interpreted as coccoid forms or Helicobacter. (Adapted from Karmeli et al. Dig Dis Sci 1995;40:2070-2073.) (N is the number of patients.)
Endoscopic Screening: Esophagogastroduodenoscopy has been used since the 1960s in Japan to detect the atrophic border,181 and to simultaneously obtain biopsies for histologic exam.238, 239 For practical reasons, endoscopic mass screening can only be recommended in gastric cancer high-incidence areas such as Japan. In Western countries, limited experience with the endoscopic appearance of gastric atrophy (using conventional white-light endoscopy or newer endoscopic techniques) and conflicting data on endoscopic biopsy sites limit endoscopic screening of at-risk patients.
Serology: Noninvasive serum biomarker tests have been used since the 1990s to screen patients for gastric atrophy.240, 241, 242 Determining serum pepsinogens, I and II (sPGI and sPGII), or sPGI:PGII ratio is an indirect measure of corpus function.243, 244, 245 PGI is secreted only by oxyntic glands. PGII is produced by all gastric glands (oxyntic, cardiac, and pyloric) as well as duodenal (Brunner’s) glands. With advancing corpus atrophy, secretion of PGI diminishes, so a low sPGI:PGII ratio is a serologic marker of corpus atrophy.240, 243, 244, 245, 246 Screening using low sPGI:PII as an atrophy marker has made it possible to screen large populations240, 241, 242 as it is convenient and economic.
Histopathology staging systems for gastric atrophy: Histology can also provide a direct measure of gastric function along with additional information about inflammation as well as the severity and topography of gastritis. A patient’s risk for gastric cancer is determined by the degree and distribution of (a) atrophy, (b) intestinal metaplasia, and (c) inflammation. Several atrophy staging systems have been designed with varying degrees of success. As the predictive value of individual biopsy specimens for cancer risk is limited, this has led to the introduction of four histopathology indices (Gastritis Risk Index,247 OLGA,248, 249 Baylor,248, 249 and Erasmus250) for gastric cancer relative risk. As each system uses different biopsy protocol systems and each has a different approach, a comparative study is of paramount importance but it is currently lacking. The various histopathology staging systems are as follows:
Gastritis Risk Index: This index uses the topographic grading of H. pylori gastritis in the antrum and corpus.118 It uses the updated Sydney system biopsy site recommendations (2 antral, 2 corpus and angulus) and its 4-point scale (negative, mild, moderate, and severe)13 to grade active and chronic inflammation. Factored into the gastritis risk index are (1) infiltration with lymphocytes and plasma cells, that is, chronic inflammation, (2) neutrophil infiltration, that is, activity, and (3) intestinal metaplasia. If chronic inflammation in the corpus is greater than or equal to that in the antrum (antrum/corpus ratio), 1 point is scored. If active inflammation in the corpus is greater than or equal to that in the antrum (antrum/corpus ratio), 1 point is again scored. If intestinal metaplasia is found in the antrum or corpus (scored as absent or present), another point is scored. The maximum score is 3 points. Using the Gastritis Index in a German population, the positive predictive value for the presence of gastric carcinoma was 46% for score 1, 79% for score 2, and 94% for score 3 compared to 17% for score 0.247 Except for intestinal metaplasia, Shimoyama et al.251 did not find a similar predictive value. Nonetheless, the control group in Shimoyama’s study included patients with atrophic gastritis.251 It is difficult to assess the reproducibility of Gastritis Risk Index as studies in other populations (claiming to use the Gastritis Risk Index) dropped the predictive value of chronic inflammation.252, 253, 254
OLGA adopts the updated Sydney’s biopsy protocol and its 4-point scale (negative, mild, moderate, and severe).248 Atrophy is defined as the loss of normal glands with and without its replacement with fibrosis, intestinal metaplasia, and pseudopyloric metaplasia.13, 255 Following the updated Sydney recommendation the final atrophy score is calculated as the average score for each region (i.e., antral atrophy is the average score of all antral biopsies [n = 3] and corpus atrophy is the average score of the two corpus biopsies). OLGA combines antral and corpus atrophy scores for a final gastric atrophy stage that relates to cancer risk248 (Table 13-7). It is noteworthy that if antral atrophy is moderate or severe (score 2 or 3) the presence of mild corpus atrophy does not increase OLGA’s score further. Though gastric cancer risk is higher in patients with extensive intestinal metaplasia, OLGA’s score equates all forms of atrophy. Elderly patients with antral fibrosis secondary to NSAID-induced ulcers will receive a high OLGA score irrespective of low cancer risk. Thus, OLGA does not take into account that gastric cancer risk increases with extensive intestinal metaplasia and with the advent of corpus atrophy.110, 120, 181 In addition, in the original study, no new patients were included as being at risk of carcinoma as a result of the study, suggesting that in practice it was of little/no value. Other investigators have suggested replacing gastric atrophy by intestinal metaplasia “OLGAIM” to increase interobserver agreement and to increase the correlation with gastritis severity.256
Table 13-7 OLGA
CORPUS ATROPHY SCORE
No atrophy (score 0)
Mild atrophy (score 1)
Moderate atrophy (score 2)
Severe atrophy (score 3)
ANTRUM ATROPHY SCORE
No atrophy (score 0)
Stage 0
Stage I
Stage II
Stage II
Mild atrophy (score 1)
Stage I
Stage I
Stage II
Stage III
Moderate atrophy (score 2)
Stage II
Stage II
Stage III
Stage IV
Severe atrophy (score 3)
Stage III
Stage III
Stage IV
Stage IV
All scores are between 0 and 3 for “mild, moderate, and severe” using a visual analogue scale.248
Baylor. Sense can be made of the issues in OLGA by following a biopsy protocol similar to Sydney’s biopsy protocol with two extra distal corpus biopsies (Fig. 13-35). Atrophy is defined as in the updated Sydney system.13 Antral and corpus atrophy stage is recorded independent of each other. Intestinal metaplasia is recorded as an independent variable. The corpus atrophy stage (early, mid, and advanced corpus atrophy) is reported on the basis of the upward extension of atrophy along the lesser and greater curvature (Fig. 13-35). Antral atrophy is recorded on a 4-point scale (negative, mild, moderate, and severe) based on degree of replacement with intestinal metaplasia. Using low sPGI:PGII as a serologic marker of corpus atrophy, Baylor’s stage showed a statistically significant inverse relationship with PGI:PGII serum levels (p < 0.0001).249
Erasmus Index uses a combination of clinical information, pepsinogen level serology, and histology to evaluate each patient’s risk for gastric cancer.250 Features factored into the Erasmus risk index are (1) family history of gastric cancer (2 points), (2) alcohol use > 1 unit/d (1 point), (3) intestinal metaplasia grade (moderate 1 point, marked 3 points), and (4) pepsinogen I to II ratio < 3 (3 points). Erasmus has a maximum score of 10; score ≥4 points was indicative of multifocal intestinal metaplasia in 96% and severe grades of intestinal metaplasia in 92%. The study examining the Erasmus Index used extensive intestinal metaplasia and not dysplasia or cancer (in follow-up biopsies) as a marker of gastric cancer risk. Dysplasia was identified in follow-up biopsies of 7 out of 32 patients (22%); all had marked intestinal metaplasia.250
Figure 13-35. The updated Sydney biopsy protocol requires a minimum of five biopsies as shown in white. These can also identify the extent of atrophy present, albeit fairly crudely, as the first site involved is the angulus/incisura (IA), and should be followed by B1, and lastly B2. It is, however, essential that each biopsy site be submitted in its own container and the site labeled for identification. By adding biopsies C1 and C2, in which atrophic changes (especially intestinal metaplasia) appear to starts at the incisura/angulus (IA), affects the antrum (A1, A2), and then extends proximally to the oxyntic zone (C1, C2), so that after IA, C1, then C2, then B1 and lastly B2 are involved biopsy site B1 is first affected.
Disorders associated withH. pylorigastritis
Peptic Ulcer Disease An ulcer is a break in the mucosa involving both the epithelium and muscularis mucosae and therefore reaches the submucosa. It therefore differs from mucosal erosions, in which the epithelial break does not penetrate the muscularis mucosae to reach the submucosa. We tend not to use the term acute ulcer, which implies a temporal or recent lesion, when outside of clinical trials this knowledge is not available, while the depth of the lesion as an ulcer rather than an erosion is also subjective. The term erosive gastritis or erosive gastropathy is preferred, recognizing that some of these lesions may penetrate the muscularis mucosae, thus qualifying in the literal sense as ulcers, but that endoscopy does not distinguish between these perfectly. Similarly, the term chronic ulcer usually implies size or depth endoscopically, but long-standing changes, for example, (fibrosis) histologically.
Peptic ulcer disease is not a single entity but a heterogeneous group of disorders having in common mucosal erosion or ulceration. PUD results when aggressive factors (acid, pepsin, bile, activated pancreatic enzymes, medications, chemicals, ischemia, radiation, etc.) overwhelm intrinsic defense mechanisms and protective factors (mucin and bicarbonate secretion and numerous systems including the cyclo-oxygenase, nitric oxide and transforming growth factor beta [TGFβ]-associated systems) (Fig. 13-36, Table 13-8). Ulcers occur primarily in the distal stomach and proximal duodenum but may occur anywhere acid and pepsin are found, for example, in the esophagus, in mucosa adjacent to surgically produced anastomoses on either side of the anastomotic line, and within ectopic gastric mucosa such as Meckel’s diverticulum. This is perhaps best illustrated with medications (NSAIDs, aspirin, etc.) that specifically overwhelm the cyclo-oxygenase system, or situations where acid is deposited directly onto an epithelium not designed to withstand it (distal to gastroenterostomy lines, gastric heterotopia in the duodenum, or Meckel’s diverticulum), and especially the Zollinger-Ellison syndrome (ZES) in which sheer volume of acid can overwhelm those parts of the intestinal tract not designed to deal with acid, and can result in other symptoms such as severe diarrhea and malabsorption.257
Figure 13-36. Imbalance between aggressive and protective factors in disease.
In practice, H. pylori infection and medication use such as NSAIDs are the most common etiologic factors for gastroduodenal ulcers.
The role of the pathologist is to exclude malignancy in the case of gastric ulcers, and to uncover causes of gastroduodenal ulceration including unusual causes (e.g., those due to Crohn’s disease [CD] or, opportunistic infection in the immunocompromised host), and to distinguish the main causes, namely, Helicobacter and medications, which is often fairly easy.
Table 13-8 Factors That Damage or Protect the Gastric Mucosa
AGGRESSIVE FACTORS
PROTECTIVE FACTORS
Endogenous
Gastric mucosal blood flow
Hydrochloric acid
Mucus
Pepsin
Bicarbonate secretion
Bile
Prostaglandins
Nitric oxide
Exogenous
Nonsteroidal anti-inflammatory drugs
Ethanol
Other
Gastroduodenal erosions and ulcers (“peptic ulcers”). In order to understand the development of ulcers, it is pertinent to discuss the mechanisms that help to maintain the integrity of the gastric mucosa. Mucosal integrity is maintained by numerous defense mechanisms, which include258
1. Pre-epithelial factors such as mucus-bicarbonate-phospholipid barrier
2. An epithelial barrier that includes surface epithelial cells connected by tight junctions and generating bicarbonate, mucus, phospholipids, trefoil peptides, prostaglandins, and heat shock proteins
3. Continuous cell renewal accomplished by proliferation of progenitor cells regulated by a variety of growth factors, for example, PGE2 and surviving TGF-β
4. An endothelial “barrier” that includes sensory innervation, generation of prostaglandins, and nitric oxide. Nitric oxide (NO) is protective and linked with NSAIDs (NO-NSAIDS), has the potential to form less injurious NSAIDs.
5. Continuous blood flow through mucosal microvessels.
Mucosal injury may occur either when noxious factors breach the intact mucosal defense system or when any of the components making up that mucosal defense is impaired. In mucosal erosions, when the surface epithelium is denuded, rapid cell restitution takes place to repair the break in the mucosa. For this to occur, intact mucosal blood flow is necessary, and prostaglandins and nitric oxide may mediate these changes. Thus an impairment of any of these factors may contribute to ulcer formation.
Patients hospitalized with severe burns have an enhanced risk of developing so-called Curling’s ulcer. Despite the clear relationship between the severity of burns and the risk of PUD, these patients secrete less acid than control subjects. In fact, there is an inverse relationship between the extent of surface burns and the rate of acid secretion. Thus, while acid is necessary for the development of PUD in burn patients and can be prevented with medications that block acid secretion, mucosal damage is not due to increased acid secretion,259 and is likely vagally mediated. In burn patients under stress, the primary factor causing ulcer disease appears to be impaired mucosal defense mechanisms against ulceration.260 It is evident that although ulcers need a milieu of acid and pepsin for their development, factors affecting mucosal resistance to injury are critically important in the development of peptic ulcers.
1.Gastric mucosal blood flow: The high metabolic rate of the gastric and duodenal epithelium requires high blood flow to maintain mucosal integrity. Mucosal ischemia is believed to be important in the pathogenesis of “stress” lesions, but whether it contributes to chronic ulcers is unknown.261, 262, 263 The role of acute ischemia in erosive gastritis is discussed subsequently.
2.Mucus: Surface columnar epithelium and mucus neck cells secrete a continuous layer of glycoprotein mucus gel (largely lost with conventional fixation and embedding264) that adheres to the mucosal surface.265 Mucus protects the underlying epithelial cells in several ways: it acts as a lubricant protecting the mucosal surface from mechanical damage by food, as a neutralizer of gastric acid, and as a barrier to luminal pepsin, thereby protecting the underlying mucosa from proteolytic digestion.266 The mucous layer has three trefoil peptides (TFFs) that have been shown to be key factors in stimulating cell migration and promoting epithelial repair after damage.267, 268, 269, 270 TFF1 is cosecreted with MUC5AC mucin in the superficial foveolar region of the stomach, TFF2 with MUC6 mucin in deep gastric and duodenal glands, and TFF3 with MUC2 mucin from goblet cells in the duodenum.267, 268, 269, 270
3.Bicarbonate secretion: Surface epithelial cells secrete bicarbonate into the adherent layer of mucus gel creating a pH gradient with a near-neutral pH at the epithelial surfaces in the stomach and duodenum. Thus the mucosal surface is in contact with neutral fluid, regardless of the amount of acid in the lumen.266 Bicarbonate secretion is depressed in the majority of peptic ulcer patients even after the ulcers have healed. Consequently, these patients have an impaired mucosal bicarbonate barrier against luminal acid and an increase in acidification at the mucosal surface.271 Secreted acid appears to go through pores in this mucous gel to reach the lumen.
4.Prostaglandins: Prostaglandins are produced by GI tract cells as well as most other tissues. Prostaglandins of the E variety are major mucosal defense mediators. Animal experiments have shown that prostaglandins can protect the gastric mucosa from damage by several noxious agents, such as aspirin, bile salts, alcohol, and boiling water.272, 273 The mechanism by which this occurs is uncertain but probably includes stimulation of mucus and bicarbonate secretion in addition to enhancing blood flow to the gastric mucosa and augmenting cell proliferation.274 Prostaglandins are responsible exclusively for maintaining mucosal integrity.275
Pathogenetic factors. Factors thought to be important in the pathogenesis of ulcer disease include genetic predisposition, increased acid and pepsin production, infections, and environmental factors, such as smoking and medications. Two important factors that disturb mucosal defense, and therefore may predispose to ulcer formation, are NSAIDS, including aspirin and H. pylori infection.116 Today, with further decline in H. pylori prevalence, other etiologic factors primarily NSAIDs play a more important role even in less developed countries.276 Indeed, we may never know the role aspirin played historically, as it was readily available prior to the rediscovery of H. pylori when it was “the” analgesic of choice until acetaminophen/paracetamol/Tylenol became available.
Decreased mucosal resistance and rapid gastric emptying are also thought to play a role (see also discussion at the beginning of this chapter).
1.Acid and pepsin. It has long been known that peptic ulcers are dependent, at least in part, on an acid/peptic milieu, giving rise to the “no acid, no ulcer” dictum stated by Karl Schwartz in 1910.277, 278, 279 Before H. pylori was rediscovered in 1984,280 it was generally accepted that duodenal ulcer was due to high acid secretion. Effective measures to reduce acid output, surgical or medical, led to long-term healing.281, 282, 283, 284 The inhibition of acid secretion heals peptic ulcers that fail to respond to conventional ulcer therapy, although when therapy is stopped, there is often rapid recurrence of ulcers, unless the initiating agent, for example, H. pylori or NSAIDs/ASA, is removed. Ulcer healing correlates with the degree and duration of acid inhibition.285 The validity of this generalization is supported by several observations. For example, peptic ulcers are exceedingly rare in atrophic gastritis associated with pernicious anemia.278, 279 Ulcers in patients with known achlorhydria strongly suggest the presence of a malignancy; patients with the most severe and intractable forms of PUD, such as the ZES/gastrinoma, have the highest rates of acid secretion.286 However, it should be noted that 50% to 60% of ulcer patients secrete normal amounts of gastric acid.287, 288, 289, 290, 291, 292, 293 While peptic damage is common, the fact that it does not occur with even greater frequency, despite the daily and nightly exposure of the stomach, distal esophagus, and duodenum to a very low pH, suggests that effective defensive factors are in place. Increased acid exposure alone is rarely sufficient to cause mucosal damage in the stomach or duodenum, although the squamous mucosa of the esophagus appears much more susceptible. The production of a gastric or duodenal ulcer at the individual level likely represents a consortium of events.
Fundic gland heterotopia, (Fig. 13-37) This refers to the presence of fundic-type glands with parietal and chief cells or commonly just parietal cells in the duodenum. This was reported to be more common (>50%) in resection specimens from duodenal ulcer patients than in nonulcer (20%) patients.294, 295 The gross findings may be those of nodules (commonly red-tipped) in the duodenal bulb. Sometimes these become very large, and ulcerate or obstruct the duodenal bulb or beyond, farther into the small bowel (see Chapter 20). Further these are known to actively secrete acid and almost certainly play a role in the genesis of duodenal ulcers. They are responsive to gastrin, and secrete acid directly into the adjacent duodenal mucosa, where the intestinal mucosa is susceptible to acid, and is frequently found at the edge of duodenal ulcers. While gastric Helicobacter clearly plays a role in duodenal ulcer disease (relative risk ×7) and especially when present in gastric surface metaplasia in the duodenum (relative risk ×51),296 the role that it plays when heterotopic mucosa is infected is unknown.
Antral parietal cells: Although infrequently discussed, it is clear that there are huge variations in the numbers of parietal cells in the antrum. In some patients they cannot be found, while in others they are numerous. These are not accompanied by chief cells unlike normal corpus glands, so PGI staining is negative in the antrum. Whether these patients are resistant to Helicobacter because the gastric pH remains low in the antrum, or conversely, when infected they have a high prevalence of duodenal ulcer because of acid secretion almost directly into the pylorus, is unknown. However, they certainly increase the gastric parietal cell mass, and sometimes actually increase in the distal antrum. Further, most pathologists are very aware of the frequency with which biopsies designated as being “antral” by our clinical colleagues consist entirely of oxyntic mucosa. This is less likely to mean they have lost their way within the stomach than that the oxyntic mucosa extends well past the angulus on the lesser curve, and much closer to the pylorus than generally believed on the greater curve. Prepyloric biopsies are therefore ideally required to obtain antral mucosa.
Hypersecretory conditions associated with peptic ulcers. In these conditions there is excessive production of gastrin, resulting in continuous stimulation of acid secretion. This includes ZES, theoretically primary gastrin cell hyperplasia (if this exists—see Chapter 5), and the retained antrum syndrome.
Zollinger-Ellison syndrome is a rare disorder (see also Chapter 5) characterized by one or more gastrin-releasing tumors (gastrinomas) in the pancreas, duodenum, or both, often in association with syndromes such as multiple endocrine neoplasia type I (MEN I). Gastrinomas release abnormal amounts of gastrin that acts as a trophic or growth-promoting hormone that induces hyperplasia of parietal and ECL cells,297, 298 resulting in excess gastric acid in the stomach and duodenum. Helicobacter pylori infection is not a major contributing factor in duodenal ulcer associated with ZES.299 In fact, H. pylori infection, as well as transient acute inflammation of the gastric mucosa, is associated with decreased acid secretion in ZES (as it may in the rest of the population).300, 301 Only a very small number of patients with PUD have ZES. Its presence should be suspected in the absence of H. pylori infection or NSAIDs usage in patients with findings suggestive of acid hypersecretion (e.g., multiple refractory ulcers, severe peptic ulcers that bleed, perforation distal to the first portion of the duodenum), diarrhea, or a personal or family history of multiple endocrine neoplasia type 1.302, 303 Morphologically the combination of parietal cells hyperplasia and ECL hyperplasia at the base of the mucosa is virtually diagnostic of this syndrome. For further details, see hypertrophic gastropathies at the end of this chapter.
Figure 13-37. A,B: Fundic gland heterotopia in the duodenal bulb. A: Low-power view. Most of the lining epithelium is of the gastric type, with the only residual villi present at the bottom right. Brunner’s glands are seen near the bottom of the section (arrow). B: Highpower view showing the interface between Brunner’s glands below and the fundic gland mucosa with parietal and chief cells above. C,D: These red bumps in the bulb represent nests of fundic gland heterotopia.
Retained antrum syndrome is an unusual complication of Billroth 2 surgery for gastric ulcer disease. In this procedure the antrum is resected, the duodenal stump is closed off, and a gastrojejunostomy is formed. If during surgery a portion of the antrum is inadvertently left behind within the duodenal stump the absence of acid in the antral remnant (and possibly duodenum where G-cells can also be found) will lead to continuous secretion of gastrin, resulting in a situation similar to gastrinoma or G-cell hyperplasia.
2.Helicobacter pylori. Helicobacter pylori infection of the stomach and duodenum is highly associated with the occurrence of peptic ulcers, especially with strains that produce the CagA.116, 304, 305, 306, 307, 308, 309 Nonetheless, some doubt H. pylori‘s role in ulcer genesis (reasons summarized in Table 13-9). In patients with duodenal ulcers, H. pylori infection induces antral gastritis, which leads to hypergastrinemia and acid hypersecretion. Excess acid enters the duodenal bulb causing or extending duodenal gastric metaplasia, allowing H. pylori colonization. This in turn induces an inflammatory response, and potentially focal erosions or ulceration. This is supported by Mongolian gerbil experiments; those infected with H. pylori develop gastritis, gastric ulcer with gastritis cystica profunda, duodenitis, gastric metaplasia, and duodenal ulcer.310 Today, the incidence of duodenal ulcers has decreased substantially in the Western world311 mirroring the fall in H. pylori infection,312 and possibly the replacement of aspirin by paracetamol/Tylenol as the analgesic of choice. In some Western countries, smoking, alcohol, and NSAIDs, but not H. pylori infection remain leading risk factors for duodenal or prepyloric ulcer.313
Table 13-9 Evidence LinkingH. pylorito the Pathogenesis of Duodenal Ulcer836, 837, 838
EVIDENCE AGAINST
EVIDENCE FOR
Craters heal, even when bacteria persist
Helicobacter pylori prevalence in DU patients varies from 85% to 100%
Acid hypersecretion can lead to DU without bacterial infection
Antibiotics heal DU at the same rate as H2-receptor antagonists or PPIs
Most infected individuals never develop DU
DU recurrence is less after antimicrobial therapy than conventional treatment for DU
If H. pylori were the primary cause, we would not see regional variation in DU prevalence within areas of high H. pylori prevalence, particularly developing countries
DU recurrence is virtually always associated with failure to clear organisms or infection relapse
Helicobacter pylori infection has been present for many centuries, but duodenal ulcer emerged only around 1900—(around the time aspirin started becoming widely available!)
Koch’s postulates have since been fulfilled for this histologic lesion
In developed countries, duodenal ulcers occur in people without H. pylori infection, raising the importance of factors such as aspirin and other NSAIDs and, much less commonly, CD
Mongolian gerbils injected with H. pylori develop duodenitis, gastric metaplasia, and duodenal ulcer
Ulcers are proportionately more common (up to 75% of all cases) in areas of low H. pylori prevalence. This may also reflect the emergence of medications as a major cause of gastroduodenal ulcers
If duodenal ulcer were predominantly (primarily) acid related, its incidence should be increasing in the Western world, in line with that of other acid-related disorders such as GERD.
Duodenal ulceration can also recur after eradication without reinfection
Successful eradication of the organism also leads to normalization of acid production by the stomach
Half of patients with acute perforations of a duodenal ulcer (i.e., with only a brief period of previous indigestion) are H. pylori-negative (medications again?)
Working hypothesis:
Working hypothesis:
Helicobacter pylori infection is secondary, delaying healing and leading to chronicity. Aspirin and NSAIDs play a huge role in what we regard as “peptic ulcer disease”
Preexisting infection with H. pylori is significantly associated with subsequent development of duodenal ulcer
3.Drugs. Millions of people throughout the world are maintained on NSAID therapy, but with the use of over-the-counter NSAIDs, and aspirin, it is almost impossible to know how many are taken. The use of NSAIDs has increased dramatically since they came into the market in the early 1970s, and has been associated with increases in the relative risk of gastric ulcers and duodenal ulcer. During a course of NSAID therapy, approximately 15% of patients will develop gastric ulcers; 5% will develop duodenal ulcers.314, 315 Several studies have shown that chronic gastric ulcers occur more frequently in patients taking large doses of aspirin compared to control populations, and the risk of NSAID-associated ulcer bleeding is higher in elderly females, probably because of the increased consumption of NSAIDs by this group.274, 316, 317, 318, 319 Interestingly, in the rare instances that gastric ulcers are resected in North America, we now find (personal experience) that chronic gastritis and H. pylori are rare, but the adjacent mucosa shows typical features of reactive gastropathy and particles of medication, presumably NSAIDs, can often be seen at the base of the ulcer (whether causative or therapeutic is a matter of debate). However, NSAID-associated upper GI bleeding does not originate from gastric ulcers alone. Duodenal ulcers may account for one-third or more of cases. Thus, although NSAIDs may only slightly increase the risk of duodenal ulcer, they appear to significantly increase the bleeding risk in those with a duodenal ulcer. Aspirin with its anticoagulant effect may potentiate this. The role that NSAIDs/ASA have played historically is outlined in the introduction to this chapter, including the association between ASA coming into the market around 1900 and the increasing numbers of patients with gastric and duodenal ulcers since that time.
The use of daily PPI therapy decreases symptoms and the development of NSAID-associated ulcers and recurrent NSAID-related ulcer complications. In patients using aspirin, the addition of a COX-2-specific inhibitor appears to significantly increase GI risk to the level of a nonselective NSAID; aspirin plus a nonselective NSAID appears to increase GI risk still higher. Patients taking low-dose aspirin who have risk factors for GI complications (including concomitant nonselective NSAID therapy) should receive medical cotherapy, such as a PPI.320
NSAID-associated ulcers are often clinically silent and probably account for about 30% of ulcer complications such as bleeding and perforation. Endoscopic erosions or ulcers are found in 15% to 30% of patients taking NSAIDS regularly, while the annual incidence of bleeding/perforation in these patients is 1% to 1.5%.258 It has been estimated that NSAIDs account for about 3,000 deaths and 20,000 hospitalizations in the United States each year.274, 317, 319, 321, 322, 323 There were over 1,000 deaths annually in elderly patients in the United Kingdom in 2000,324 so the figure in the United States may well be many times this.325 Indeed, one estimate suggested 16,500 deaths from GI-related causes in 1997 from NSAIDs use in the United States.326 However, the rates are likely lower now for various reasons including the concurrent use of PPI and also Coxibs.
Only gold members can continue reading. Log In or Register to continue
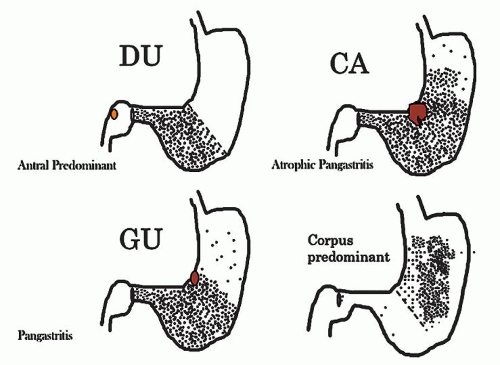


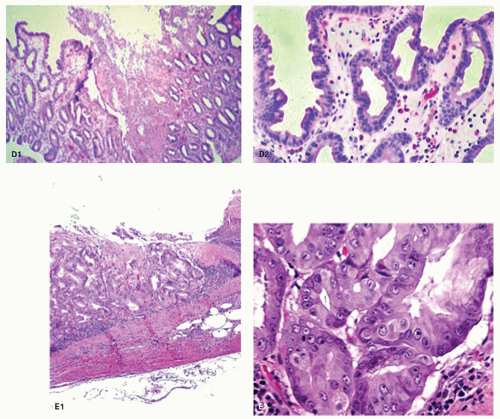
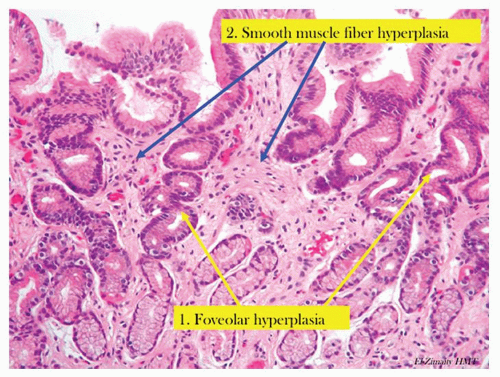

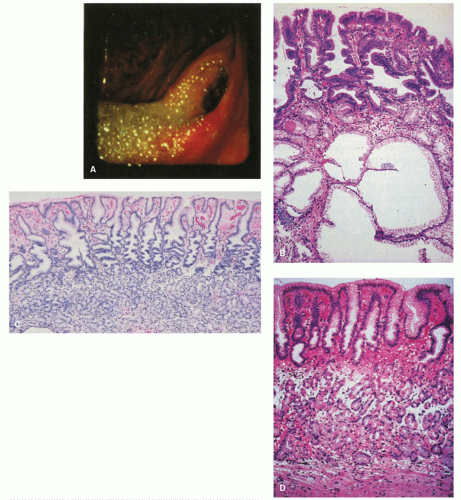
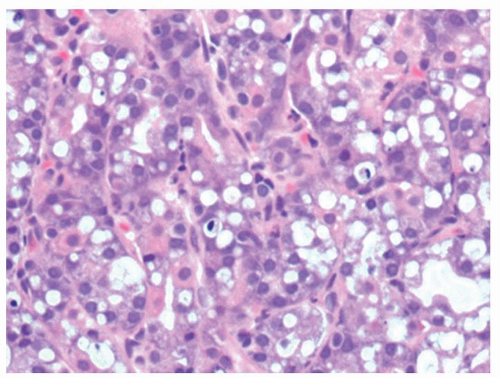
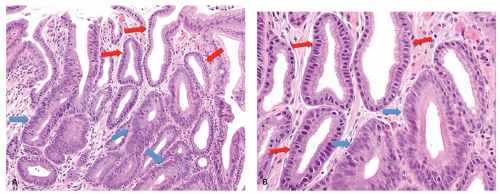

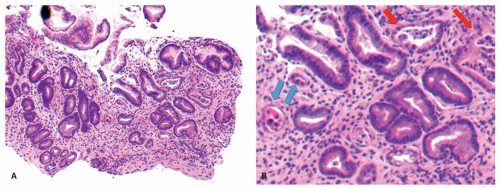

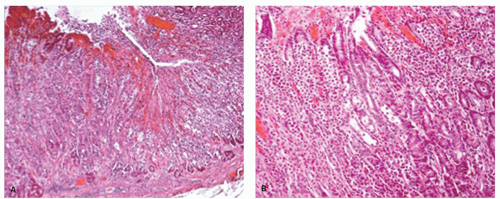

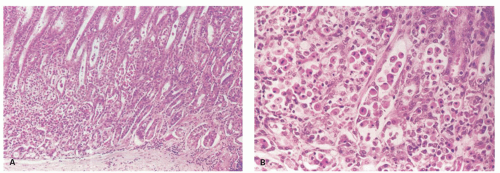

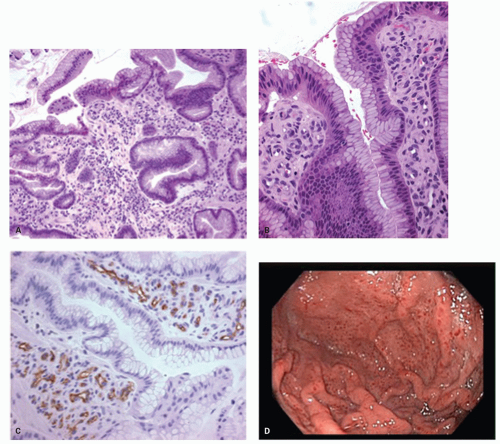
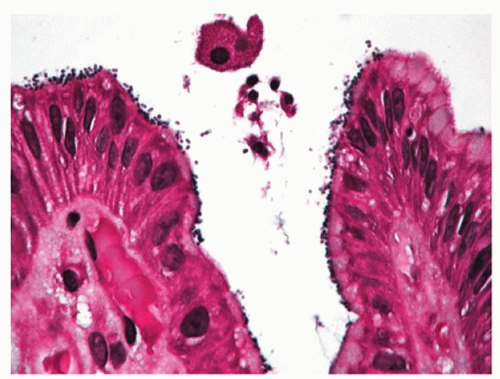
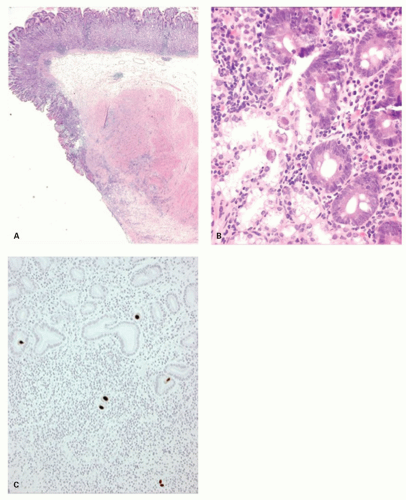
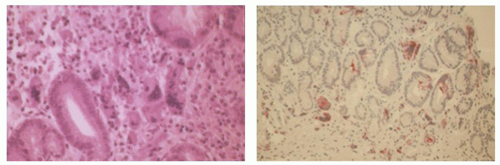
 ) and CO2 in a thin neutral layer around the outer surface of the bacteria.123 The ammonium ions markedly increase the pH in its surrounding environment to neutral (or above), which is necessary for its survival.124 Helicobacter pylori further protects itself by swimming through the protective layer of gastric mucus away from the acidic contents of the lumen toward the more neutral pH environment of the epithelial cells beneath.125 Nonetheless, H. pylori flourishes
) and CO2 in a thin neutral layer around the outer surface of the bacteria.123 The ammonium ions markedly increase the pH in its surrounding environment to neutral (or above), which is necessary for its survival.124 Helicobacter pylori further protects itself by swimming through the protective layer of gastric mucus away from the acidic contents of the lumen toward the more neutral pH environment of the epithelial cells beneath.125 Nonetheless, H. pylori flourishes