Central obesity is involved in the pathogenesis and progression of Barrett’s esophagus to esophageal adenocarcinoma. Involved are likely both mechanical and nonmechanical effects. Mechanical effects of increased abdominal fat cause disruption of the gastroesophageal reflux barrier leading to increased reflux events. Nonmechanical effects may be mediated by inflammation, via classically activated macrophages, pro-inflammatory cytokines, and adipokines such as Leptin, all of which likely potentiate reflux-mediated inflammation. Insulin resistance, associated with central obesity, is also associated with both Barrett’s pathogenesis and progression to adenocarcinoma. Molecular pathways activated in obesity, inflammation and insulin resistance overlap with those involved in Barrett’s pathogenesis and progression.
Key points
- •
Central obesity is an independent risk factor for Barrett’s esophagus (BE) and esophageal adenocarcinoma. Overall body fat content is not a risk factor for BE.
- •
Central obesity likely mediates its influence on BE via mechanical and metabolic effects.
- •
Mechanical effects include disruption of the gastroesophageal junction reflux barrier leading to some increase in gastroesophageal reflux.
- •
Metabolic effects are likely mediated by the visceral abdominal fat compartment, which releases adipokines, and leads to a systemic inflammatory state (mediated via the release of saturated free fatty acids, macrophage activation, and release of pro-inflammatory cytokines), resulting in insulin resistance.
- •
Several of these inflammatory and proneoplastic pathways can be potentially targeted by novel molecular therapies for chemoprevention.
Introduction
An exponential increase in the prevalence of obesity has been observed in the United States in recent decades, paralleled by a comparable increase in the incidence of esophageal adenocarcinoma (EAC), a cancer that continues to carry a bleak prognosis. These unprecedented increases in obesity and EAC assume particular importance in the context of Barrett’s esophagus (BE), the only established premalignant precursor and strongest risk factor of EAC. In BE, the stratified squamous lining of the distal esophagus undergoes a metaplastic change to an intestinal-type columnar epithelium, presumably due to long-standing exposure to gastric refluxate. BE may progress via the development of dysplasia to EAC. The origin of BE and mechanisms of progression to EAC, however, are poorly understood. An obesity and reflux-driven synergism could potentially explain some aspects of the pathogenesis and progression of BE. This review summarizes the current evidence on the role of obesity in the development and progression of BE to EAC.
Introduction
An exponential increase in the prevalence of obesity has been observed in the United States in recent decades, paralleled by a comparable increase in the incidence of esophageal adenocarcinoma (EAC), a cancer that continues to carry a bleak prognosis. These unprecedented increases in obesity and EAC assume particular importance in the context of Barrett’s esophagus (BE), the only established premalignant precursor and strongest risk factor of EAC. In BE, the stratified squamous lining of the distal esophagus undergoes a metaplastic change to an intestinal-type columnar epithelium, presumably due to long-standing exposure to gastric refluxate. BE may progress via the development of dysplasia to EAC. The origin of BE and mechanisms of progression to EAC, however, are poorly understood. An obesity and reflux-driven synergism could potentially explain some aspects of the pathogenesis and progression of BE. This review summarizes the current evidence on the role of obesity in the development and progression of BE to EAC.
Obesity and Barrett’s esophagus: evidence for association
Obesity can have wide-ranging consequences on the human body, both metabolic and nonmetabolic, but its association with BE is particularly intriguing. Traditionally, association studies of obesity and BE have used the body mass index (BMI), a convenient, but imprecise tool to measure overall body fat mass. Meta-analyses of observational evidence by Cook et al. and Kamat et al. have shown an association between BMI and BE when using population-based controls without gastroesophageal reflux disease (GERD), but their analyses suffered from the limitation of using unadjusted odds ratios (OR).A GERD-adjusted analysis by Cook showed that the association between BMI and BE was no longer significant when using GERD controls, leading the investigators to speculate that BMI was a risk factor for BE, but with the association mediated through GERD. Given that BMI does not accurately measure body fat content, the findings from studies of association of BMI with BE, or the lack thereof, are not surprising. More recently, researchers have tried to use other measurement tools, such as DEXA (dual-energy X-ray absorptiometry) and bioelectrical impedance analysis (BIA) that can capture body fat mass more precisely. Thrift and colleagues conducted a case-control study in which body fat mass was measured by BIA. They found that BMI was not associated with an increased risk of BE, despite using this more accurate method to measure overall body fat mass.
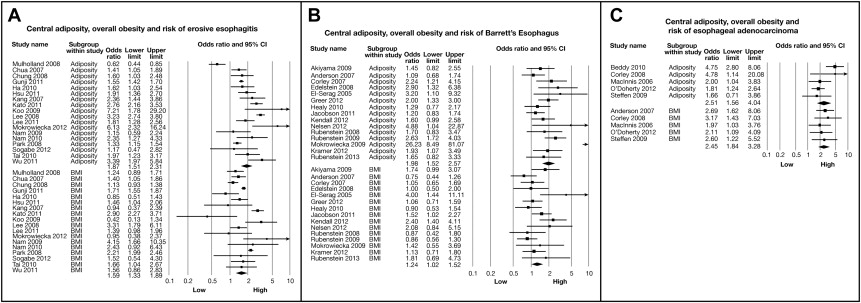
In contrast to BMI, central obesity has a more robust association with BE. A hospital-based case-control study by Kramer et al. comparing 237 BE cases with 1021 endoscopy controls showed that waist-to-hip ratio (WHR) was associated with a nearly 2-fold increase in BE risk, independent of GERD; this association was further strengthened when the analysis was limited to long-segment Barrett’s esophagus (LSBE) and white men. Thrift and colleagues found a similar association between WHR and BE in a case-control study using primary care and endoscopy controls, but the association lost significance when the analysis was adjusted for GERD. Another study found all measures of central obesity (waist circumference [WC], WHR, sagittal abdominal diameter, and waist-to-height ratio) to be strongly associated with increased risk of BE in men, but not in women; this association was reduced slightly after adjusting for GERD symptoms. A recent meta-analysis of 40 observational studies has conclusively shown that central adiposity (measured by either anthropometry or quantitative assessment of intra-abdominal fat: subcutaneous and visceral, by computed tomography [CT]) is associated with an increased risk of BE, even after BMI and GERD are controlled for ; the effect estimate was more pronounced in LSBE patients and in men ( Fig. 1 ). A dose-response relationship further strengthened this association. This association was seen only with visceral fat and not subcutaneous fat. This association was in addition to strong associations with erosive esophagitis and EAC, phenotypes that are closely linked to BE. An additional meta-analysis using individual patient data from the BEACON consortium has shown that increased WC is associated with a greater than 2-fold higher risk of BE in both men and women, independent of BMI and GERD symptoms.
Obesity and Barrett’s esophagus: mechanisms of association
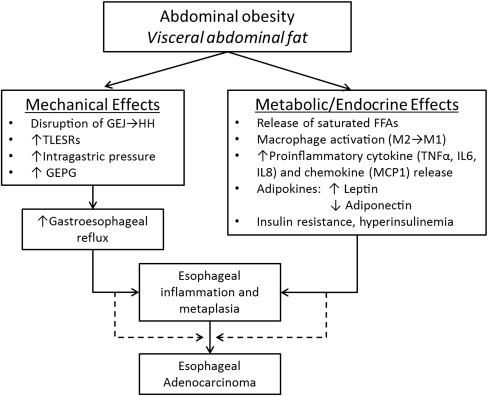
Ample evidence suggests that abdominal obesity is involved in the pathogenesis of BE. Abdominal obesity is presumably thought to have a dual effect in BE pathogenesis and progression: one that is GERD-dependent (mechanical effect), and another that is GERD-independent (nonmechanical or metabolic effect). Abdominal fat is mainly distributed in 2 compartments: a subcutaneous fat compartment and a visceral fat compartment. Studies that have measured visceral fat using radiographic techniques, such as single-slice abdominal CT (at the level of the L2-L3 interspace), demonstrate that it is indeed the visceral portion of the abdominal fat that is associated with an increased risk of BE, and not subcutaneous fat. Although both subcutaneous and visceral fat likely exert mechanical effects, visceral fat is more likely responsible for the metabolic effect. These 2 mechanisms are examined in the following discussion ( Fig. 2 ).
Mechanical Effects of Central Obesity
Central obesity, by exerting mechanical effects on the gastroesophageal junction (GEJ), likely results in its anatomic disruption. A prospective study of 285 patients using high-resolution manometry showed that central obesity, measured by WC, had a strong positive correlation with raised intragastric pressure and moderate positive correlation with gastroesophageal pressure gradient (GEPG), after controlling for age, sex, and gastroesophageal reflux symptoms. A dose-response relationship between increasing WC and increased intragastric pressure and GEPG was also observed. Furthermore, both BMI and WC were shown to be associated with anatomic disruption of the GEJ (separation of the diaphragmatic hiatus and lower esophageal sphincter), which leads to the formation of a hiatal hernia. These findings have largely been confirmed by Derakhshan et al. in a sample of dyspeptic patients having normal endoscopy (without evidence of a hiatal hernia or erosive esophagitis).
In a Chinese population-based study largely without GERD symptoms, Wu and colleagues found that when compared with controls, a greater proportion of obese patients had an increased frequency of postprandial transient lower esophageal sphincter relaxations (TLESR) with acid reflux, which is another crucial physiologic mechanism of gastroesophageal reflux. The study also demonstrated a significantly increased postprandial GEPG in obese patients. Both BMI and WC were found to correlate well with their findings. Another cross-sectional study involving 206 patients showed a modest association between obesity (BMI ≥30 and increased WC) and increased postprandial acid reflux in those who were obese when compared to normal subjects. However, when both BMI and WC (as a measure of central obesity) were examined together, BMI was found to have no association with esophageal acid exposure, indicating that central obesity was likely mediating most of the effects of obesity on GERD. Indeed, meta-analyses have shown that both hiatal hernia and GERD are associated with BE.
Despite these reported findings, the mechanical effect alone is insufficient to explain the association of abdominal obesity with BE. For instance, in a prospective cross-sectional study involving 322 patients, there was only a weak positive correlation between intragastric pressure and measures of obesity (both BMI and WC) after adjusting for age, gender, race, and indications for manometry. The absolute increase in reflux episodes with obesity is modest. In addition, other gastrointestinal cancers, which are not influenced by reflux (such as colon and pancreas ), are also independently associated with central obesity. Perhaps the most compelling evidence is provided by the meta-analysis by Singh et al., where, even after adjusting for GERD, the association between abdominal obesity and BE still persisted, indicating that this association might be mediated by GERD independent of nonmechanical effects (see Fig. 1 ). Hence, central obesity, although likely increasing mechanical pressure leading to some increase in gastric reflux, also contributes to BE and EAC risk by nonmechanical means.
Metabolic Effects of Central Obesity
Visceral abdominal fat and obesity
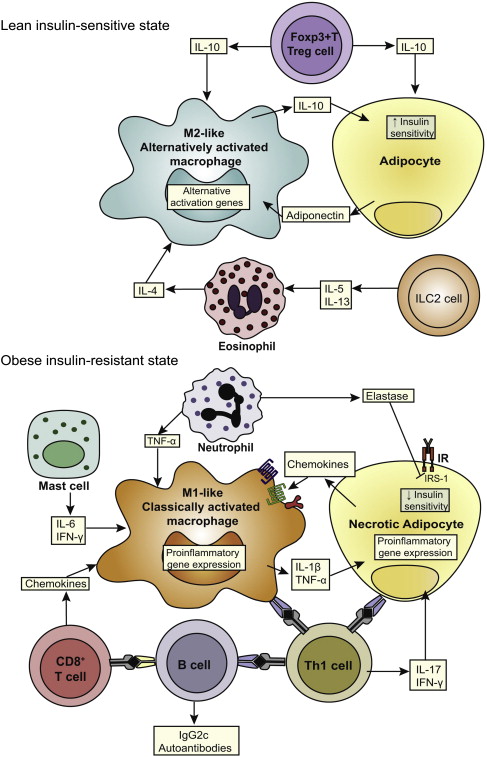
Obesity is widely recognized as a state of chronic low-grade systemic inflammation. Visceral adipose tissue contains an abundance of pro-inflammatory cells, such as classically activated (M1) macrophages, neutrophils, mast cells, Th1 (CD4+, CD8+) T lymphocytes, and B lymphocytes as well as anti-inflammatory cells, such as alternatively activated (M2) macrophages, T regulatory and Th2 lymphocytes, and eosinophils ( Fig. 3 ). In normal physiologic states (lean individuals), the overall balance between these cells remains anti-inflammatory, preventing the overexpression of inflammatory mediators. However, in the obese state, adipocyte hypertrophy along with tissue hypoxia leads to a breakdown of this homeostasis. Pro-inflammatory cells, particularly classically activated macrophages (recruited by chemokines such as macrophage chemoattractant protein 1), increase, leading to overexpression and systemic release of pro-inflammatory cytokines (tumor necrosis factor-α [TNF-α], interleukin (IL)-1β, IL-6), lipolysis (which leads to the release of saturated free fatty acids [FFAs]), and a change in the pattern of hormones released by adipocytes (adipokines, such as leptin: increased levels, and adiponectin: decreased levels). Saturated FFA released by lipolysis also promote activation of macrophages to an M1 phenotype. These obesity induced changes cumulatively lead to a systemic inflammatory state and insulin resistance, both of which have been associated with an increased risk of BE and EAC.
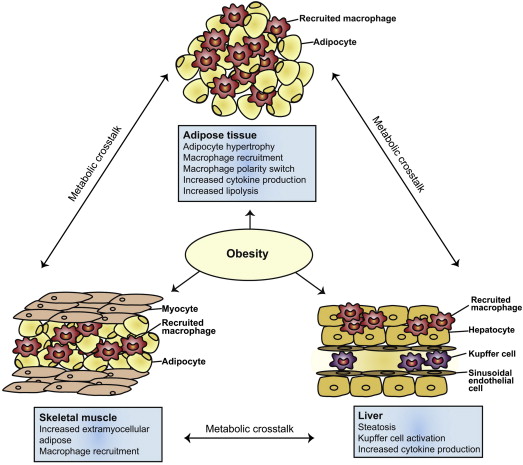
In addition to visceral adipose tissue, obesity has been shown to be associated with macrophage-mediated inflammation in other organs, such as the liver, pancreatic islets, hypothalamus, and skeletal muscle ( Fig. 4 ). Hence, obesity may lead to both systemic and localized organ level inflammation in several target organs. Both of these mechanisms may be operative in enhancing esophageal injury and inflammation. Increased peri-esophageal GEJ fat has been demonstrated in subjects with BE and correlates with histologic inflammation and high-grade dysplasia (HGD) ; this may enhance esophageal inflammation by paracrine mechanisms. In addition, increased macrophage infiltration has been demonstrated in the BE epithelium of centrally obese BE subjects compared with controls. Most macrophages in the BE epithelium appear to be of a pro-inflammatory (M1) phenotype, which could enhance reflux-mediated inflammation.
Leptin and Barrett’s esophagus
Serum leptin, a product of the ( ob ) gene, is proportional to adipose tissue mass. Leptin levels are substantially higher in women than in men, even after accounting for total body fat. Leptin is angiogenic and mitogenic and regulates neovascularization. It is also anti-apoptotic. Binding of leptin to its receptor results initiates the phosphorylation of JAK2 tyrosine kinase, which in turns starts a cellular signaling cascade that results in further downstream activation of extracellular signal-regulated kinases (ERK), Akt, nuclear factor κB (NFkB), p38, and JNK, all of which are necessary for leptin to exert its anti-apoptotic and proliferative effects. It has been shown that the esophageal mucosa is densely packed with leptin receptors. Hence, it is hypothesized that in the obese state, leptin, acting synergistically with stomach acid or alone, induces proliferation in the distal esophagus.
Epidemiologic studies have looked at the association between increased serum leptin and BE. In a GERD-adjusted analysis comparing 306 BE cases and 309 controls, Kendall and colleagues found a 2-fold higher risk of BE when serum leptin levels were high, but only in men. Rubenstein et al. found a similar significant association between high serum leptin and BE risk in men after adjusting for obesity, GERD, and serum insulin. Garcia and colleagues found that high levels of leptin were associated with an 8-fold greater risk of BE in cases than in controls; this association remained significant even after excluding women from the analysis. Thompson et al., on the contrary, found a greater than 3-fold risk of association between increased serum leptin and BE, but only in women. The reasons for this discrepancy are unclear, but could be related to populations studied, assays used to measure leptin levels, and other unmeasured/unadjusted confounders like insulin, which was adjusted for in only one study. In a recent meta-analysis of 6 studies reporting on the association of serum leptin with BE, Devanna and colleagues found that increased serum leptin was associated with a 2-fold increased risk of BE. This association was strengthened when the analysis was adjusted for GERD, suggesting an independent role of leptin in BE pathogenesis.
Adiponectin and Barrett’s esophagus
Adiponectin is a 30-kDa polypeptide secreted predominantly by visceral adipose tissue. Its levels in serum are inversely proportional to the amount of body fat and are lower in men than women. In contrast to leptin, adiponectin exhibits anti-inflammatory and anti-angiogeneic properties. Adiponectin also antagonizes the effect of leptin by inhibiting leptin-induced cellular proliferation. Three adiponectin multimers are recognized in circulating blood, namely, low-molecular-weight (LMW), middle-molecular-weight, and high-molecular-weight (HMW) adiponectin. HMW and LMW adiponectin multimers are known to have contrasting pro-inflammatory and anti-inflammatory actions, respectively.
Two adiponectin receptors are recognized in tissues, namely Adipo-R1 and R2. These receptors have been found to be decreased in BE cell lines. In cell lines, adiponectin inhibits progression of BE into EAC by exerting strong anti-apoptotic effects. BE proliferation is linked to an abnormal activation of the ERK1/2 in the squamous mucosa. Physiologically, high serum adiponectin levels suppress these kinases. In obesity, as a result of low adiponectin, unchecked cellular proliferation takes place.
Mokrowiecka and colleagues found significantly lower serum adiponectin levels in BE cases when compared with control patients. In another study, Rubenstein et al. found an inverse association between low total serum adiponectin and BE after adjusting for GERD, smoking, and WHR. However, the study was limited by its small sample size and only measured total serum adiponectin and not its multimers. In a second larger study, the same investigators looked at the differential effects of the 3 multimeric forms of adiponectin. Comparing 112 BE cases and 199 GERD controls, they found high levels of LMW adiponectin to be associated with a significantly lower risk of BE. When stratified by gender, this association became stronger in women, but not in men. The association remained after adjusting for potential confounding by insulin/insulin resistance. Thompson and colleagues also showed an inverse relationship between high levels of total adiponectin and BE after adjusting for age, tobacco use, leptin, and WHR; this association was lost when the analysis was stratified by gender. Two other observational studies found no association between serum adiponectin and BE. A systematic review and meta-analysis failed to demonstrate any association between total serum adiponectin and risk of BE. Despite inconclusive results from epidemiologic studies, the study by Rubenstein informs us that the physiologic actions of adiponectin are probably mediated by its LMW multimer. By upregulating anti-inflammatory cytokines and downregulating pro-inflammatory cytokines, LMW adiponectin is thought to prevent inflammatory damage to the squamous epithelium in the setting of GERD and perhaps impedes the development and proliferation of BE. Barrett’s epithelium shows greater expression of the pro-inflammatory cytokine IL-6, adding strength to this postulation.
Insulin and insulin resistance
It is now well accepted that obesity is one of the principal contributory factors in the development of insulin resistance (IR). Systemic inflammation associated with obesity has been shown to be a crucial mechanism responsible for IR. Insulin resistance is a condition where peripheral tissues are not sensitive to insulin despite high circulating insulin levels. Insulin stimulates the production of insulin-like growth factor (IGF-1) and downregulates production of IGFBP1, resulting in higher levels of IGF-1 in serum. IGF-1 binds to its receptor (IGF-1R) and this leads to activation of the PI3K/AKT/mTOR pathway, which subsequently promotes mitogenesis and prevents apoptosis. Such an activation of the IGF pathway has been shown to occur in BE. Furthermore, molecular studies have shown that IGF-1 and its binding proteins could influence neoplastic progression in BE. Insulin and insulin-related signaling pathways have been shown to be upregulated in BE and EAC tissue. IR is also associated with dysplastic progression in BE, adding further proof that obesity-induced hyperinsulinemia and IR are associated with BE pathogenesis and progression.
Obesity also upregulates several inflammatory pathways, such as NFkB and JNK, which are known to play a role in the development of IR. Overexpression of B cells, which occurs in obesity, is also associated with IR. In addition to inflammatory mediators, obesity-induced IR also seems to be mediated by adipokines. Leptin and adiponectin have opposite effects on insulin and IR; elevated serum leptin is strongly associated with IR, whereas high adiponectin increases peripheral tissue sensitivity to insulin. Low adiponectin levels have been implicated in obesity-induced IR.
Several observational studies have looked at the association of insulin, IR, and IGF with BE. Levels of insulin and the pro-inflammatory cytokine, IL-6, were found to be higher in BE cases than controls in a cross-sectional study by Ryan and colleagues. In a large population-based case-control study, type 2 diabetes mellitus was associated with a nearly 50% increase in the risk of BE, independent of other known risk factors such as GERD and BMI; this association was stronger in women than men. Leggett found a 2-fold increase in the risk of BE in those with metabolic syndrome (which is associated with insulin resistance) that was also independent of BMI and GERD. Greer et al. found a strong relationship between increased insulin and IGF-1 levels and risk of BE, but only when compared with healthy controls; this association disappeared when the comparison was GERD controls, indicating that the relationship between insulin and BE could be GERD-dependent. Rubenstein and colleagues, on the other hand, could not find an association between increased insulin levels and higher risk of BE, when controlling for serum leptin. Two other case-control studies similarly failed to show any association between increased insulin and BE. There could be several reasons for this nonsignificant result in some studies. Effects on the esophageal mucosa may not be directly mediated by insulin or IR, rather through leptin and/or other adipokines. Insulin could exert its effect through GERD, which has been shown to be increased in hyperinsulinemic and IR states. Hence, adjusting for GERD (which was done in most studies) could have attenuated or removed any significant association between insulin and BE.
Related posts:
 Forthcoming Issues
Predictors of Progression to High-Grade Dysplasia or Adenocarcinoma in Barrett’s Esophagus
Forthcoming Issues
Predictors of Progression to High-Grade Dysplasia or Adenocarcinoma in Barrett’s Esophagus
 Advanced Imaging in Barrett’s Esophagus
Screening for Barrett’s Esophagus
Biomarkers in Barrett’s Esophagus
Genetic and Epigenetic Alterations in Barrett’s Esophagus and Esophageal Adenocarcinoma
Advanced Imaging in Barrett’s Esophagus
Screening for Barrett’s Esophagus
Biomarkers in Barrett’s Esophagus
Genetic and Epigenetic Alterations in Barrett’s Esophagus and Esophageal Adenocarcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


