The optimal approach to vitamin D supplementation for the average healthy person is debatable. In patients with cancer, the role of vitamin D supplementation, possibly in treatment, is even less clear. Vitamin D is shown to play a role in prostate cancer biology; however, the clinical data have not consistently demonstrated a link. Additional studies are needed to determine if higher doses of vitamin D supplements could benefit selected populations (ie, the elderly or patients with cancer) even if they may not be beneficial for the general population.
At present, the optimal approach to vitamin D supplementation for the average healthy person is debated. In patients with cancer, the role of vitamin D supplementation in treatment, is even less clear. With the recent publication of new guidelines by the Institute of Medicine (IOM) for vitamin D supplementation, the question how much vitamin D a human requires arises. For years, many physicians have been recommending vitamin D supplementation at doses somewhat higher than national recommendations because evidence had suggested that vitamin D may play a role in the prevention and treatment of several diseases. The hypothesis that this evidence suggests has been of significant interest to cancer researchers.
For decades, there have been efforts to test the hypothesis that low vitamin D plays a role in prostate cancer development and progression. Epidemiologic observations show that populations in areas with low UV exposure have an increased risk of prostate cancer, and laboratory experiments showing antitumor effects of vitamin D have provided evidence supporting this hypothesis and have stimulated further studies examining the relationship between prostate cancer and vitamin D in humans. However, studies looking for a direct association between serum vitamin D levels and prostate cancer have not consistently supported this hypothesis. In addition, the use of vitamin D as a therapeutic addition to prostate cancer treatments has not achieved consistently positive results to date.
Vitamin D biology
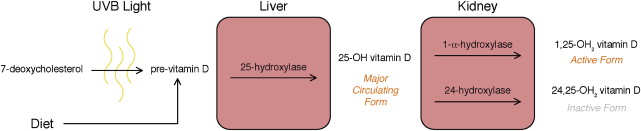
Vitamin D acts as a regulatory hormone for multiple cell activities in the human body. The activated form of vitamin D is 1,25-OH 2 vitamin D, which is formed through several tightly regulated synthesis steps ( Fig. 1 ). The process begins with 7-deoxycholesterol that is converted to pre–vitamin D in the presence of UV-B light. The 7-deoxycholesterol is then converted to 25-OH vitamin D, the main circulating form of vitamin D in the body, by the enzyme 25-hydroxylase, predominantly in the liver. The final conversion to 1,25-OH 2 vitamin D, which is catalyzed by 1α-hydroxylase, primarily occurs in the kidney but can also be seen in other tissues such as the prostate, which can potentially play a role in cancer pathogenesis.
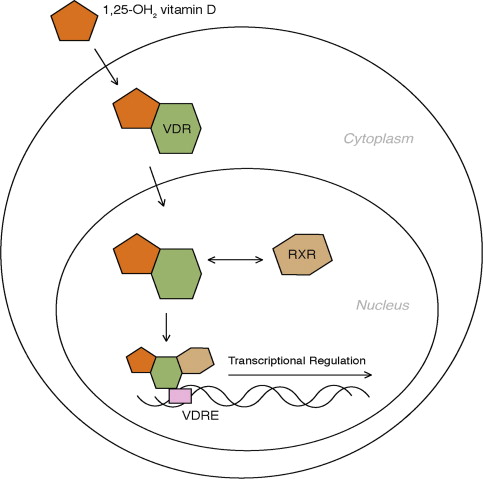
The vitamin D receptor (VDR) is a steroid receptor. When bound by 1,25-OH 2 vitamin D, the VDR is translocated to the nucleus to regulate gene expression ( Fig. 2 ). In this way, the VDR acts as a ligand-activated (1,25-OH 2 vitamin D) transcription factor. Once activated, the VDR forms a heterodimer with retinoid-X receptor, which aids in binding the receptor complex to DNA. The activated VDR then binds to the promoter region of specific genes with vitamin D response elements (VDREs) and regulates the transcription of messenger RNA in these genes. The regulatory effect of vitamin D on calcium through transcriptional regulation of genes such as osteocalcin and PTH is well established, but additional genes with VDREs are increasingly being discovered. For example, cell cycle regulators, such as p21, Bcl-2, and insulinlike growth factor, and cell signaling molecules, such as tumor necrosis factor α and epidermal growth factor receptor, have been found to have VDREs. The transcriptional regulation, either upregulation or downregulation, of these genes is the basis for the observed experimental antineoplastic effects of vitamin D. Nongenomic direct actions of the VDR on cell signaling and growth have also been described.
Vitamin D anti–prostate cancer activity
With many cell cycle regulatory genes having VDREs, multiple studies have focused on examining the potential antitumor activity of vitamin D in different tumor models. There have been multiple examples of vitamin D–induced cell cycle arrest in G1 and inhibition of mitosis induction pathways. There have also been VDRs demonstrated on prostate carcinoma cell lines. In cell lines, cell culture, and mouse models, vitamin D has been shown to have antiproliferative and, therefore, antineoplastic actions.
A question that arises is why have these antineoplastic effects of vitamin D not been consistently seen with prostate cancer in human experience, although they have been shown in the laboratory. One hypothesis is based on the ability of prostate cells to locally activate 1,25-OH 2 vitamin D from 25-OH vitamin D through the enzymatic activity of 1α-hydroxylase.
Normal prostate cells have been found to express 1α-hydroxylase. As mentioned, most activations to 1,25-OH 2 vitamin D occur in the kidney, so why would prostate cells need to express their own 1α-hydroxylase? Perhaps the ability of the prostate cell to locally activate vitamin D is important for normal cell regulation. It has been shown that prostate cancer cells lose the activity of 1α-hydroxlase in the laboratory. However, this phenomenon has not been fully characterized in humans. Loss of local 1α-hydroxylase may render prostate cancer cells more dependent on circulating 1,25-OH 2 vitamin D rather than on the more abundant precursor 25-OH vitamin D, which can reduce the prostate cell’s ability to regulate its own growth in response to vitamin D levels. To demonstrate this finding, restoring 1α-hydroxylase activity in LNCaP cells with gene transfer has been shown to restore the antineoplastic effects of 25-OH vitamin D.
Because 1,25-OH 2 vitamin D levels are usually tightly regulated and only decrease during severe deficiency states, tissues that rely on renally activated vitamin D are relatively unaffected by mild vitamin D deficiency states. However, tissues, such as the prostate, that may rely on local production of 1,25-OH 2 vitamin D could experience decreased VDR signaling with small changes in 25-OH vitamin D levels, which are more closely linked to a patient’s overall vitamin D status. The decreased 1α-hydroxlase activity in prostate cancer cells should render the cells more free to proliferate in states without adequate circulating 25-OH vitamin D levels. This finding is yet to be demonstrated in human studies measuring 25-OH vitamin D serum levels, and it is therefore not known if the changes in local vitamin D synthetic capacity observed in models of prostate carcinogenesis are important in the human disease.
Vitamin D anti–prostate cancer activity
With many cell cycle regulatory genes having VDREs, multiple studies have focused on examining the potential antitumor activity of vitamin D in different tumor models. There have been multiple examples of vitamin D–induced cell cycle arrest in G1 and inhibition of mitosis induction pathways. There have also been VDRs demonstrated on prostate carcinoma cell lines. In cell lines, cell culture, and mouse models, vitamin D has been shown to have antiproliferative and, therefore, antineoplastic actions.
A question that arises is why have these antineoplastic effects of vitamin D not been consistently seen with prostate cancer in human experience, although they have been shown in the laboratory. One hypothesis is based on the ability of prostate cells to locally activate 1,25-OH 2 vitamin D from 25-OH vitamin D through the enzymatic activity of 1α-hydroxylase.
Normal prostate cells have been found to express 1α-hydroxylase. As mentioned, most activations to 1,25-OH 2 vitamin D occur in the kidney, so why would prostate cells need to express their own 1α-hydroxylase? Perhaps the ability of the prostate cell to locally activate vitamin D is important for normal cell regulation. It has been shown that prostate cancer cells lose the activity of 1α-hydroxlase in the laboratory. However, this phenomenon has not been fully characterized in humans. Loss of local 1α-hydroxylase may render prostate cancer cells more dependent on circulating 1,25-OH 2 vitamin D rather than on the more abundant precursor 25-OH vitamin D, which can reduce the prostate cell’s ability to regulate its own growth in response to vitamin D levels. To demonstrate this finding, restoring 1α-hydroxylase activity in LNCaP cells with gene transfer has been shown to restore the antineoplastic effects of 25-OH vitamin D.
Because 1,25-OH 2 vitamin D levels are usually tightly regulated and only decrease during severe deficiency states, tissues that rely on renally activated vitamin D are relatively unaffected by mild vitamin D deficiency states. However, tissues, such as the prostate, that may rely on local production of 1,25-OH 2 vitamin D could experience decreased VDR signaling with small changes in 25-OH vitamin D levels, which are more closely linked to a patient’s overall vitamin D status. The decreased 1α-hydroxlase activity in prostate cancer cells should render the cells more free to proliferate in states without adequate circulating 25-OH vitamin D levels. This finding is yet to be demonstrated in human studies measuring 25-OH vitamin D serum levels, and it is therefore not known if the changes in local vitamin D synthetic capacity observed in models of prostate carcinogenesis are important in the human disease.
Prostate cancer and vitamin D: epidemiologic evidence
There are multiple laboratory studies showing that vitamin D can play a role in prostate cancer biology, but what evidence is there in humans to confirm that vitamin D can have an appreciable effect on prostate cancer development or progression?
UV Exposure Data
The hypothesis that vitamin D may play a role in cancer biology was formed after geographic studies demonstrated that there may be an increased risk of dying from cancer in areas with low UV exposure. This geographic distribution of cancer-related mortality, with the highest risk in northern regions, was seen in a variety of cancers, including prostate cancer ( Table 1 ). The initial studies, from the early 1990s, have been confirmed in later geographic studies as well. In addition to an increased prostate cancer mortality based on geographic location, cancer mortality risk has also been shown to have seasonal variations. Two recent studies have shown that there is a decreased risk of prostate cancer death when a patient is diagnosed in the summer, both showing a relative risk reduction of approximately 20%.
| Study | Year | Location | Measure | If Low UV Exposure is Associated with Increased Prostate Cancer Risk or Mortality |
|---|---|---|---|---|
| Hanchette | 1992 | United States | Scored locations in the United States based on a combination of cloud cover, latitude, and altitude | Yes, increased mortality |
| Luscombe | 2001 | United Kingdom | Sunbathing score | Yes, increased risk |
| Grant | 2002 | United States | Scored locations in the United States based on UV-B radiation map | Yes, increased mortality |
| Bodiwala | 2003 | United Kingdom | Sunbathing score | Yes, increased risk |
| John | 2005 | Western United States | Measured skin pigmentation | Yes, increased risk (of advanced prostate cancer) |
| Schwartz | 2006 | United States | UV index data | Yes, increased mortality |
| Robsahm | 2004 | Norway | Scored regions in Norway based on latitude and climate | No a |
| Lagunova | 2007 | Norway | Annual ambient UV exposure and incidence of squamous cell carcinoma of the skin | No b |
a Diagnosis of prostate cancer in autumn was associated with decreased mortality (relative risk, 0.83).
b Diagnosis of prostate cancer in summer and autumn was associated with decreased mortality (relative risk, 0.8).
Of the 8 studies performed that examined markers of UV exposure, an impressive 75% returned a result linking reduced UV exposure to increased prostate cancer risk or increased prostate cancer–related mortality. Further, the 2 studies that did not show a geographically linked increased prostate cancer risk did show a difference in mortality for season of diagnosis.
Dietary and Supplement Data
There are multiple feedback loops that regulate the activation of 1,25-OH 2 vitamin D. One of these feedback loops includes high serum calcium levels acting to reduce the activity of 1α-hydroxylase, which in turn reduces the synthesis of active 1,25-OH 2 vitamin D, thus decreasing the absorption of calcium. With this in mind, it would be expected that high calcium intake could have a detrimental effect on active vitamin D production and, thus, an increased risk of prostate cancer if the UV exposure hypothesis is correct. However, this finding has not been shown consistently across multiple studies. There have been 12 recent studies comparing calcium and vitamin D intake and the risk of developing prostate cancer ( Table 2 ). Of these studies, 8 showed no association with calcium intake (variously defined) and prostate cancer risk. Of those eight studies, 2 showed an association with one form of calcium intake and prostate cancer risk but not another. One found an association with total calcium and dietary intake but not with supplemental calcium intake. One study even showed a decreased risk of prostate cancer with higher calcium intake. There were, however, 6 studies that showed some increased prostate cancer risk with increased calcium intake. These results could be explained by differences in calcium absorption. A study that had measured serum calcium levels to clarify this result showed an increased risk of death caused by prostate cancer with higher serum calcium levels. Serum calcium level, however, is not a strong indicator of calcium intake because it reflects the homeostatic balance of calcium exchange within the bone matrix, renal calcium handling, as well as intake and absorption. None of these 12 studies showed a link between vitamin D intake and prostate cancer risk.
| Study | Year | Number of Subjects | Age (y) | Diet/Supplementation | Is There Association with Prostate Cancer Risk |
|---|---|---|---|---|---|
| Chan | 1998 | 1062 | <80 | Calcium and dairy intake | Yes, increased risk |
| Giovannucci | 1998 | 1792 | 40–75 | Dietary and supplementary calcium | Yes, increased risk of aggressive and metastatic prostate cancer |
| Skinner | 2008 | 2814 | 24–77 | Serum calcium | Yes, increased risk |
| Kristal | 2010 | 9559 | >55 | Calcium intake | Yes, decreased risk of high-grade prostate cancer |
| Vitamin D intake | No | ||||
| Ahn | 2007 | 29,509 | 55–74 | Dietary calcium | Yes, increased risk |
| Supplemental calcium | No | ||||
| Rodriguez | 2003 | 65,321 | 50–74 | Total calcium intake | Yes, increased risk |
| Dairy intake | No | ||||
| Kristal | 2002 | 605 | 40–64 | Calcium intake | Yes, increased risk of aggressive prostate cancer |
| Vitamin D intake | No | ||||
| Park | 2007 | 10,180 | 50–71 | Calcium intake | No |
| Chan | 2000 | 27,111 | 50–69 | Calcium intake | No |
| Baron , a | 2005 | 672 | 61.8 (mean) | Calcium supplementation | No |
| Berndt | 2002 | 454 | 46–92 | Calcium intake | No |
| Tavani | 2005 | 2745 | 46–74 | Calcium intake | No |
| Vitamin D intake | No | ||||
| Tavani | 2001 | 1050 | 45–79 | Calcium intake | No |
Related posts:
 Quercetin for Chronic Prostatitis/Chronic Pelvic Pain Syndrome
Quercetin for Chronic Prostatitis/Chronic Pelvic Pain Syndrome
 The Complex Interplay Between Cholesterol and Prostate Malignancy
Pollen Extract for Chronic Prostatitis—Chronic Pelvic Pain Syndrome
Molecular Markers that Can Be Utilized in Diet and Dietary Supplement Research
Statin Clinical Trial (REALITY) for Prostate Cancer: an Over 15-Year Wait is Finally Over Thanks to a Dietary Supplement
Heart Health = Urologic Health and Heart Unhealthy = Urologic Unhealthy: Rapid Review of Lifestyle Changes and Dietary Supplements
The Complex Interplay Between Cholesterol and Prostate Malignancy
Pollen Extract for Chronic Prostatitis—Chronic Pelvic Pain Syndrome
Molecular Markers that Can Be Utilized in Diet and Dietary Supplement Research
Statin Clinical Trial (REALITY) for Prostate Cancer: an Over 15-Year Wait is Finally Over Thanks to a Dietary Supplement
Heart Health = Urologic Health and Heart Unhealthy = Urologic Unhealthy: Rapid Review of Lifestyle Changes and Dietary Supplements
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


