CHAPTER 50 Pediatric Oncology
![]() What is the most common renal tumor of infancy, and which renal tumor is most common during childhood?
What is the most common renal tumor of infancy, and which renal tumor is most common during childhood?
Congenital mesoblastic nephroma is the most common tumor in infants, while Wilms is the most common tumor in children. After the age of 10, renal tumors in children are just as likely to represent a Wilms tumor or a pediatric renal cell carcinoma.
![]() What is the most common extracranial solid tumor of childhood?
What is the most common extracranial solid tumor of childhood?
Neuroblastoma is the most common extracranial solid tumor in children. It accounts for 6% to 10% of all childhood malignancies.
![]() The Wilms tumor suppressor gene, WT-1, is located on which chromosome?
The Wilms tumor suppressor gene, WT-1, is located on which chromosome?
The Wilms tumor suppressor gene (WT-1) is located on the distal portion of the short arm of chromosome 11 in the 11p13 location. A second putative Wilms tumor gene has been found at 11p15 (WT-2). Abnormalities at this location are associated with overgrowth syndrome and Beckwith–Weidemann syndrome.
![]() What congenital anomalies are associated with Wilms tumor, and what chromosomal abnormalities are associated with these anomalies?
What congenital anomalies are associated with Wilms tumor, and what chromosomal abnormalities are associated with these anomalies?
Congenital anomalies are seen in approximately 15% of patients with Wilms tumor. These include the following:
• WT-1:
Aniridia.
WAGR syndrome (Wilms, Aniridia, Genitourinary anomalies, and Retardation)—tumor risk 30%.
• Hemihypertrophy:
Denys–Drash syndrome (Wilms, pseudohermaphroditism, glomerulopathy, retardation)—tumor risk 90%.
• WT-2:
Beckwith–Wiedemann syndrome (macroglossia, gigantism, and organomegaly)—tumor risk 5%.
• Xq26:
Simpson–Golabi–Behmel syndrome (organomegaly, renal dysplasia, polydactyly, cardiac defects)—tumor risk 7.5%.
![]() What is the significance of loss of heterozygosity (LOH) at sites 16q and 1p in Wilms tumor?
What is the significance of loss of heterozygosity (LOH) at sites 16q and 1p in Wilms tumor?
LOH at 16q (seen in 20% of all tumors) and 1p (seen in 10% of all tumors) have been identified as biomarkers of worse outcome. When occurring together, they represent an indication for more aggressive therapy.
![]() The histologic picture of Wilms tumor is described as “triphasic.” What are these 3 components?
The histologic picture of Wilms tumor is described as “triphasic.” What are these 3 components?
The classic microscopic appearance of Wilms tumor includes blastemal, epithelial, and stromal components.
![]() What is the significance of nephrogenic rests?
What is the significance of nephrogenic rests?
More than one third of kidneys resected for Wilms tumor contain precursor lesions, known as nephrogenic rests. Nephrogenic rests can be separated into 2 distinct categories: perilobar nephrogenic rests and intralobar nephrogenic rests. These 2 types of rests are distinguished by their location within the renal lobe. Perilobar nephrogenic rests are found only in the lobar periphery, which is elaborated late in embryogenesis, while intralobar nephrogenic rests are found anywhere within the lobe, as well as the renal sinus and the wall of the pelvicalyceal system.
![]() What is the significance of multiple nephrogenic rests within a single kidney?
What is the significance of multiple nephrogenic rests within a single kidney?
Multiple rests in one kidney usually implies that nephrogenic rests are present in the other kidney.
![]() What is the significance of nephrogenic rests in Wilms tumor patients who are less than 12 months old?
What is the significance of nephrogenic rests in Wilms tumor patients who are less than 12 months old?
Children less than 12 months of age who are diagnosed with Wilms tumor and also have rests, particularly perilobar nephrogenic rests, have a markedly increased risk of developing contralateral disease and require frequent and regular surveillance for several years. Surveillance is also recommended for those diagnosed after 12 months of age who have nephrogenic rests.
![]() What are the current recommendations regarding screening for Wilms tumor?
What are the current recommendations regarding screening for Wilms tumor?
Screening with serial renal ultrasonography has been recommended in children at high risk for development of Wilms tumor, including those with WAGR syndrome, hemihypertrophy, Denys–Drash syndrome, or Beckwith–Wiedemann syndrome. Review of most studies suggests that 3 to 4 months is the appropriate screening interval. Tumors detected by screening will generally be lower stage. Screening of the contralateral kidney following nephrectomy for unilateral Wilms tumor is also recommended. CT or MRI should be performed if ultrasonography demonstrates a suspicious lesion.
![]() How should regional lymph nodes be addressed during surgery for Wilms tumor?
How should regional lymph nodes be addressed during surgery for Wilms tumor?
Selective sampling of lymph nodes is an essential component of local tumor staging and is required by children’s oncology group (COG) guidelines. Formal retroperitoneal lymph node dissection (RPLND) is not recommended. Extensive lymph node dissection, particularly above the renal hilum, can result in chylous ascites.
![]() A patient with Wilms tumor undergoes nephrectomy. The tumor penetrates the surface of the capsule. At the time of nephrectomy, there is tumor spillage, which is confined to the flank. Lymph nodes are negative. Contralateral kidney is negative. Chest x-ray is negative. What stage is this tumor?
A patient with Wilms tumor undergoes nephrectomy. The tumor penetrates the surface of the capsule. At the time of nephrectomy, there is tumor spillage, which is confined to the flank. Lymph nodes are negative. Contralateral kidney is negative. Chest x-ray is negative. What stage is this tumor?
This is a stage III Wilms tumor. The staging system is as follows:
Stage I: Tumor limited to kidney, completely resected. The renal capsule is intact. The tumor was not ruptured or biopsied prior to removal. The vessels of the renal sinus are not involved. There is no evidence of tumor at or beyond the margins of resection.
Note: For a tumor to qualify for certain therapeutic protocols as stage I, regional lymph nodes must be examined microscopically.
Stage II: The tumor is completely resected, and there is no evidence of tumor at or beyond the margins of resection. The tumor extends beyond the kidney, as is evidenced by any one of the following criteria:
• There is regional extension of the tumor (ie, penetration of the renal capsule, or extensive invasion of the soft tissue of the renal sinus, as discussed below).
• Blood vessels within the nephrectomy specimen outside the renal parenchyma, including those of the renal sinus, contain tumor.
Note: Tumor spillage confined to the flank, including biopsy of the tumor, is no longer included in stage II and is now included in stage III.
Stage III: Residual nonhematogenous tumor present following surgery and confined to abdomen. Any one of the following may occur:
• Lymph nodes within the abdomen or pelvis are involved by tumor. (Lymph node involvement in the thorax or other extra-abdominal sites is a criterion for stage IV.)
• The tumor has penetrated through the peritoneal surface.
• Tumor implants are found on the peritoneal surface.
• Gross or microscopic tumor remains postoperatively (eg, tumor cells are found at the margin of surgical resection on microscopic examination).
• The tumor is not completely resectable because of local infiltration into vital structures.
• Tumor spillage occurring either before or during surgery.
• The tumor was biopsied (whether tru-cut, open, or fine needle aspiration) before removal.
• Tumor is removed in greater than one piece (eg, tumor cells are found in a separately excised adrenal gland; a tumor thrombus within the renal vein is removed separately from the nephrectomy specimen).
Stage IV: Hematogenous metastases (lung, liver, bone, brain, etc), or lymph node metastases outside the abdomino-pelvic region are present. (The presence of tumor within the adrenal gland is not interpreted as metastasis, and staging depends on all other staging parameters present.)
Stage V: Bilateral renal involvement by tumor is present at diagnosis. An attempt should be made to stage each side according to the above criteria on the basis of the extent of disease.
![]() What hematologic abnormality has been associated with patients with Wilms tumor?
What hematologic abnormality has been associated with patients with Wilms tumor?
Acquired von Willebrand disease has been found in 8% of newly diagnosed Wilms tumor patients.
![]() What is the most important factor in the prognosis of patients with Wilms tumor?
What is the most important factor in the prognosis of patients with Wilms tumor?
The most important aspect of Wilms tumor is the histology. Unfavorable histology (anaplasia) at any stage is associated with a worse prognosis. Anaplasia is seen in approximately 10% of all Wilms tumors. Anaplasia is defined by the presence of gigantic polyploid nuclei within the tumor sample. It is rare in the first 2 years of life, but the incidence increases to 13% in children older than 5 years old. Anaplasia is further divided into focal and diffuse forms, with diffuse anaplasia carrying a worse prognosis than focal. Anaplasia is believed to be a marker of chemoresistance.
![]() When should partial nephrectomy be considered for Wilms tumor?
When should partial nephrectomy be considered for Wilms tumor?
Partial nephrectomy for Wilms tumor should be considered in patients with a solitary kidney, bilateral tumors, and in those with congenital anomalies predisposing them to Wilms tumor development.
![]() How often do synchronous bilateral Wilms tumors occur, and what is the management approach?
How often do synchronous bilateral Wilms tumors occur, and what is the management approach?
Synchronous bilateral Wilms tumors occur in 5% to 7% of children with Wilms tumor. Children with bilateral tumors should not undergo initial radical nephrectomy. These children should receive preoperative chemotherapy with the goal of tumor shrinkage and renal-sparing surgery. Biopsy is not needed if the radiographic picture is consistent with Wilms tumor. Tumor response is assessed after 6 weeks with CT or MRI to determine the reduction in tumor volume and feasibility of partial resection. Tumors not responding to therapy require bilateral open biopsy to determine histology. Open biopsies are recommended, because they are more accurate than percutaneous needle biopsies when assessing for anaplasia. Failure to achieve a reduction in volume is likely due to tumor differentiation. All patients should undergo definitive surgery by 12 weeks.
![]() What is the current survival in Wilms tumor?
What is the current survival in Wilms tumor?
Data from NWTS IV indicate that for patients with favorable histology and unilateral tumors stages I to IV, 10-year overall survival ranged from 96% to 81%, respectively. For patients with anaplastic tumors stages II and III, survival was 49%, and stage IV patients with anaplasia had a survival rate of 18% at 10 years.
![]() Which patients with Wilms tumor are classified as very low risk, and how does their management differ?
Which patients with Wilms tumor are classified as very low risk, and how does their management differ?
Children less than 2 years of age with Stage I favorable histology Wilms tumors <550 g are classified as very low risk. They have an excellent prognosis when treated with nephrectomy alone without adjuvant chemotherapy.
![]() An 18-month-old male presents with malaise, weight loss, and diffuse pain. An abdominal mass is palpable. What is the most likely diagnosis?
An 18-month-old male presents with malaise, weight loss, and diffuse pain. An abdominal mass is palpable. What is the most likely diagnosis?
Neuroblastoma. At the time of presentation of neuroblastoma, approximately 70% of patients have disseminated disease versus less than 10% to 15% with Wilms. Patients with Wilms tumor generally appear in good health, in contrast to patients with neuroblastoma in whom the tumor may be widespread at presentation, and systemic symptoms are often present.
![]() Congenital neuroblastoma can mimic what other tumor?
Congenital neuroblastoma can mimic what other tumor?
Congenital neuroblastoma may present in a similar fashion as pheochromocytoma, with symptoms produced by secretion of catecholamines (hypertension, tachycardia).
![]() When does the peak incidence of neuroblastoma occur?
When does the peak incidence of neuroblastoma occur?
The peak incidence of neuroblastoma is seen in the first year of life. Median age at diagnosis is 19 months. The incidence decreases progressively with age, with 36% occurring in infants, 89% in children younger than 5 years, and 98% diagnosed by 10 years of age.
![]() Neuroblastoma is a malignant neuroendocrine tumor. There is a spectrum ranging from neuroblastoma to other benign tumors. What are these other tumors, and what are their characteristics?
Neuroblastoma is a malignant neuroendocrine tumor. There is a spectrum ranging from neuroblastoma to other benign tumors. What are these other tumors, and what are their characteristics?
Ganglioneuroma is the benign counterpart to neuroblastoma. It does not metastasize, but it can envelop adjacent structures and even extend into intervertebral foramina. Histologically, it contains mature ganglion cells in a collagen-rich background. Ganglioneuromas are most often diagnosed in older children and are usually located in the posterior mediastinum and retroperitoneum, with only a small number arising in the adrenal glands.
Ganglioneuroblastoma is intermediate between neuroblastoma and ganglioneuroma. There is a histologic spectrum between these; the outcome for ganglioneuroblastoma is varied.
![]() What is the most common site of occurrence of neuroblastomas?
What is the most common site of occurrence of neuroblastomas?
Seventy-five percent originate in the abdomen (retroperitoneum). Fifty percent arise in the adrenal, and 25% occur in the paravertebral ganglia.
![]() Is age a significant determinant of outcome in neuroblastoma?
Is age a significant determinant of outcome in neuroblastoma?
Yes, age less than 1 year is an indicator of better outcome when compared to older children, likely due to more favorable biology in tumors occurring at a younger age. Other predictive factors include site of origin, with better survival noted for nonadrenal primary tumors. Stage of disease is also an important predictor, as stage I and II patients have excellent survival rates.
![]() When is a bone marrow aspirate and/or biopsy indicated in the evaluation of patients with confirmed or suspected neuroblastoma?
When is a bone marrow aspirate and/or biopsy indicated in the evaluation of patients with confirmed or suspected neuroblastoma?
All patients with neuroblastoma should undergo bone marrow aspirate and/or biopsy. As many as 70% of bone marrow aspirates have been reported to be positive in patients with neuroblastoma.
![]() What molecular markers have been shown to carry prognostic significance in patients with neuroblastoma?
What molecular markers have been shown to carry prognostic significance in patients with neuroblastoma?
• N-myc amplification is associated with a poor prognosis and is found in about 30% to 40% of patients with advanced disease.
• Hyperdiploid tumors (those that contain more than the normal amount of DNA material found in somatic cells) have demonstrated a favorable outcome.
![]() What are the characteristics of stage IV-S neuroblastoma?
What are the characteristics of stage IV-S neuroblastoma?
Stage IV-S describes patients who would otherwise be classified with stage I (tumor confined to the organ of origin) or stage II (tumor extending in continuity beyond the organ of origin, but not crossing the midline; ipsilateral regional nodes may be involved but contralateral nodes must be negative) but who have remote spread of tumor confined to one or more of the following: liver, skin, or bone marrow (without evidence of bony involvement). This stage is restricted to children with less than 10% bone marrow involvement and to patients younger than 1 year. This stage accounts for 7% to 10% of all cases of neuroblastoma. This group typically has an excellent prognosis, with survival rate in excess of 80%. Many of these tumors undergo spontaneous regression.
![]() What diagnostic tests can be useful for making the diagnosis of neuroblastoma, and what specific CT findings would help distinguish neuroblastoma from Wilms tumor?
What diagnostic tests can be useful for making the diagnosis of neuroblastoma, and what specific CT findings would help distinguish neuroblastoma from Wilms tumor?
• Urinary metabolites of catecholamines. Vanillylmandelic acid (VMA) and homovanillic acid (HVA) are found to be elevated in 90% to 95% of patients.
• Anemia is associated with diffuse bone marrow involvement.
• The finding of intratumoral calcifications, vascular encasement, or both on preoperative computed tomography may help distinguish neuroblastoma from Wilms tumor.
• In addition to standard radiologic imaging tests, metaiodobenzylguanidine (MIBG) scintigraphy can be useful in identifying primary and metastatic lesions.
![]() What is the age distribution among patients with rhabdomyosarcoma?
What is the age distribution among patients with rhabdomyosarcoma?
A bimodal age distribution exists, whereby peaks in incidence of rhabdomyosarcoma occur in children between the ages of 0 and 2 and then again later in adolescence.
![]() What are the histologic types of rhabdomyosarcoma, and which is most common in childhood?
What are the histologic types of rhabdomyosarcoma, and which is most common in childhood?
The types of rhabdomyosarcoma are embryonal, alveolar, and undifferentiated. Mixed rhabdomyosarcomas also occur, and in about 10% to 20% of cases, the tumor is so undifferentiated that it does not fit into the standard classification.
Embryonal rhabdomyosarcoma is the most common type seen in childhood, accounting for two thirds of genitourinary (GU) rhabdomyosarcomas, and 50% to 70% of all rhabdomyosarcomas in children.
Sarcoma botryoides is a descriptive term for an exophytic embryonal tumor that looks grossly like a cluster of grapes. It tends to arise in hollow organs (bladder, vagina).
Another histologic variant of the embryonal type is the “spindle cell sarcoma.” It often arises in a paratesticular location and is associated with an especially favorable outcome.
The second most common form is alveolar, comprising 15% to 20% of cases. Alveolar rhabdomyosarcoma (ARMS) occurs more commonly in the trunk and extremities than in genitourinary sites. ARMS is associated with translocations t(2; 13) and t(1; 13). Translocation fusion positive tumors comprise 80% of ARMS and are more aggressive. Fusion-negative ARMS may have a clinical course similar to embryonal rhabdomyosarcoma.
The third category consists of undifferentiated tumors that also fare poorly.
![]() What are the most common GU sites?
What are the most common GU sites?
Prostate, bladder, paratesticular appendages, followed by the vagina and uterus.
![]() If a patient has a post-treatment biopsy indicating mature rhabdomyoblasts, should the patient have exonerative surgery to remove these elements?
If a patient has a post-treatment biopsy indicating mature rhabdomyoblasts, should the patient have exonerative surgery to remove these elements?
No, current evidence suggests that rhabdomyoblasts alone on biopsy after treatment does not require aggressive surgical treatment. Observation alone is needed.
![]() Besides histologic type, what are some adverse prognostic factors in patients with rhabdomyosarcoma?
Besides histologic type, what are some adverse prognostic factors in patients with rhabdomyosarcoma?
Tumors arising in the prostate, children younger than 1 year, and tumors in adults and older children generally have a poorer prognosis.
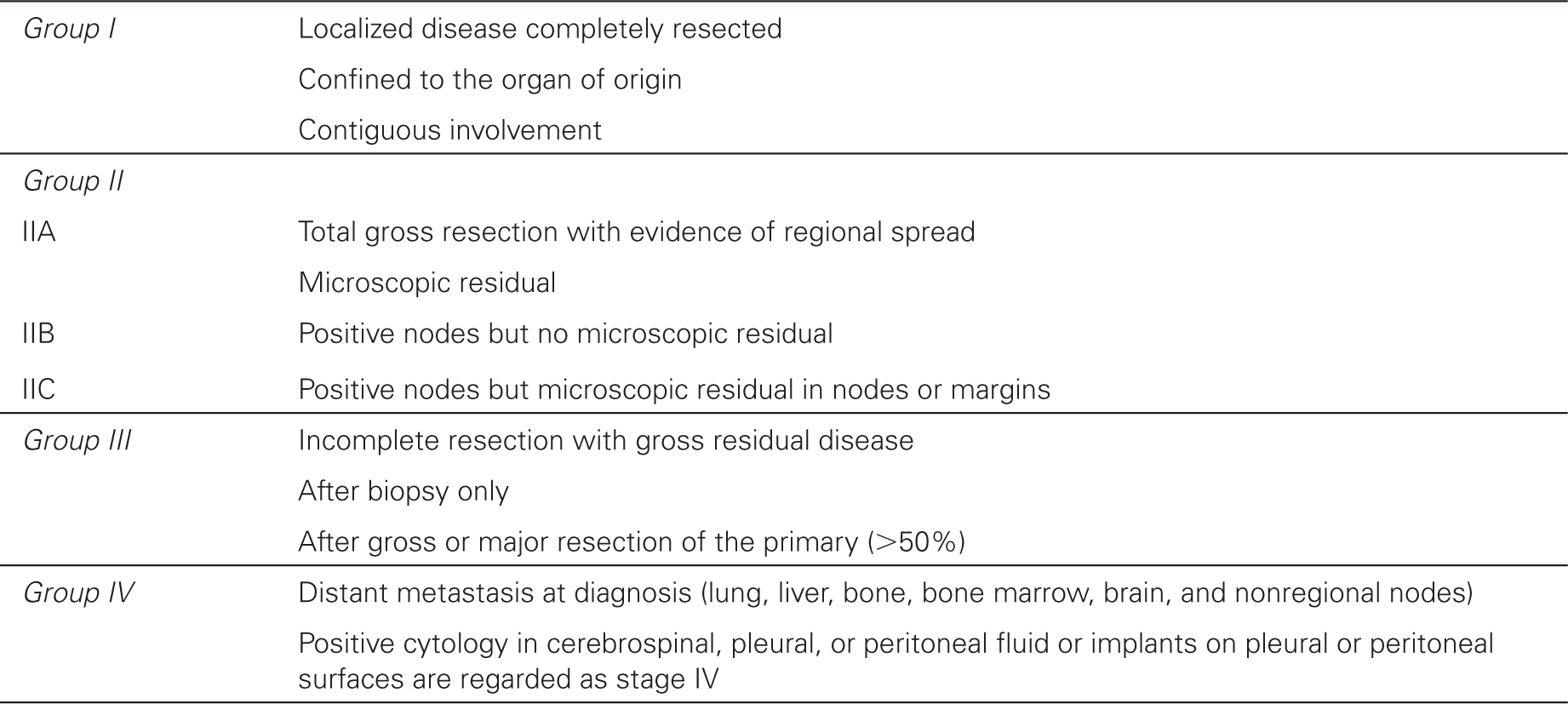
![]() A 4-year-old male has a rhabdomyosarcoma of the bladder. At the time of surgery, the tumor appears grossly removed. However, pathological analysis reveals positive margins. Lymph nodes are negative. According to the International Rhabdomyosarcoma Study (IRS) staging system, what stage disease is this?
A 4-year-old male has a rhabdomyosarcoma of the bladder. At the time of surgery, the tumor appears grossly removed. However, pathological analysis reveals positive margins. Lymph nodes are negative. According to the International Rhabdomyosarcoma Study (IRS) staging system, what stage disease is this?
This patient is in group IIA. The staging system is as follows:
Intergroup Rhabdomyosarcoma Study Group Clinical Grouping Classification
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


