Chapter 23 Pediatric liver disease
1 The liver is physiologically immature during the perinatal period, and significant maturational changes in hepatic metabolic processes occur during childhood; these metabolic processes affect the presentation of and reaction to viral and toxin exposures.
2 Acquired liver diseases that are seen in adults are rare in children. More commonly encountered in children are congenital or metabolic disorders.
3 Liver disease in children may manifest as hyperbilirubinemia, hepatomegaly, liver failure, cirrhosis, cystic disease of the liver, portal hypertension, or systemic disease resulting from the secondary effects of liver disease.





Consequences of Physiologic Immaturity of the Liver
1. Altered metabolism and clearance of potentially toxic endogenous and exogenous toxic compounds:
 Hepatic concentrations of cytochrome P-450 are low in infants. Similarly, activities of aminopyrine N-demethylase and aniline p-hydroxylase are low. Hepatic processes, such as clearance of certain drugs or bilirubin that depend on these systems, are inefficient. Therefore, potentially toxic serum levels of such compounds may be reached more rapidly in infants than in older persons.
Hepatic concentrations of cytochrome P-450 are low in infants. Similarly, activities of aminopyrine N-demethylase and aniline p-hydroxylase are low. Hepatic processes, such as clearance of certain drugs or bilirubin that depend on these systems, are inefficient. Therefore, potentially toxic serum levels of such compounds may be reached more rapidly in infants than in older persons.
Hepatic concentrations of cytochrome P-450 are low in infants. Similarly, activities of aminopyrine N-demethylase and aniline p-hydroxylase are low. Hepatic processes, such as clearance of certain drugs or bilirubin that depend on these systems, are inefficient. Therefore, potentially toxic serum levels of such compounds may be reached more rapidly in infants than in older persons.

2. Bile acid pool size and composition may be altered. It is unclear whether the alterations are beneficial (some of the bile acids present may be more readily excreted in urine) or harmful (the atypical bile acids that are formed may exacerbate cholestasis).
3. Physiologic jaundice: Up to one third of newborns develop unconjugated hyperbilirubinemia within the first week of life. Breast-fed infants have a higher risk of developing jaundice than do formula-fed infants. This occurrence is often referred to as physiologic jaundice and resolves spontaneously with no complications. Physiologic jaundice reflects the transition from clearance and metabolism of unconjugated bilirubin by the maternal system to that of the infant. The pathogenesis is likely multifactorial:
 Increased production of bilirubin: The newborn has a large red cell mass, and the cells have a shorter half-life than adult red cells.
Increased production of bilirubin: The newborn has a large red cell mass, and the cells have a shorter half-life than adult red cells.
Increased production of bilirubin: The newborn has a large red cell mass, and the cells have a shorter half-life than adult red cells.




Hyperbilirubinemia
Pathophysiologic Mechanisms
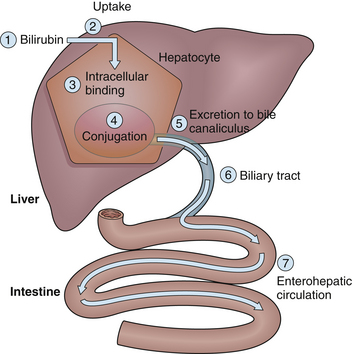
Alterations in any step of bilirubin metabolism may cause jaundice in excess of physiologic jaundice (Fig. 23.1; the numbers in the figure correspond to the numbers (1–7) in the following outline):
1. Increased bilirubin production: This can result from an increase in the release of heme from red blood cells, for the following reasons:
 Hemolysis due to Rh incompatibility, ABO incompatibility, or other minor blood group incompatibilities
Hemolysis due to Rh incompatibility, ABO incompatibility, or other minor blood group incompatibilities
Hemolysis due to Rh incompatibility, ABO incompatibility, or other minor blood group incompatibilities

2. Decreased bilirubin uptake into the hepatocyte:
 This may be caused by hypothyroidism or gestational hormones that may inhibit the uptake of bilirubin across the hepatocyte membrane. Thyroxine is important for liver plasma membrane function.
This may be caused by hypothyroidism or gestational hormones that may inhibit the uptake of bilirubin across the hepatocyte membrane. Thyroxine is important for liver plasma membrane function.
This may be caused by hypothyroidism or gestational hormones that may inhibit the uptake of bilirubin across the hepatocyte membrane. Thyroxine is important for liver plasma membrane function.
3. Abnormalities of intracellular binding or storage of bilirubin within hepatocytes: These are rare disorders and include deficiencies or alteration in GST, the primary intracellular binding protein for bilirubin.

4. Inefficient conjugation of bilirubin within the hepatocyte: Within the hepatocyte, bilirubin is conjugated with glucuronic acid by bilirubin uridine diphosphate (UDP)-glucuronyl transferase to form bilirubin monoglucuronide or diglucuronide.
 A decrease in UDP-glucuronyl transferase activity is seen in Gilbert’s syndrome, resulting in benign elevations in serum unconjugated bilirubin levels, especially during stresses such as viral illnesses.
A decrease in UDP-glucuronyl transferase activity is seen in Gilbert’s syndrome, resulting in benign elevations in serum unconjugated bilirubin levels, especially during stresses such as viral illnesses.
A decrease in UDP-glucuronyl transferase activity is seen in Gilbert’s syndrome, resulting in benign elevations in serum unconjugated bilirubin levels, especially during stresses such as viral illnesses.

5. Alterations in the excretion of bilirubin through the canalicular membrane into the biliary tract: Bilirubin diglucuronide is excreted into the canaliculus by a carrier protein.
 Alterations in this carrier protein are thought to be the cause of Dubin–Johnson syndrome. The genetic defect is on chromosome 10q24. Although this syndrome has no associated morbidity or mortality, it is characterized by elevated serum levels of conjugated and unconjugated serum bilirubin.
Alterations in this carrier protein are thought to be the cause of Dubin–Johnson syndrome. The genetic defect is on chromosome 10q24. Although this syndrome has no associated morbidity or mortality, it is characterized by elevated serum levels of conjugated and unconjugated serum bilirubin.
Alterations in this carrier protein are thought to be the cause of Dubin–Johnson syndrome. The genetic defect is on chromosome 10q24. Although this syndrome has no associated morbidity or mortality, it is characterized by elevated serum levels of conjugated and unconjugated serum bilirubin.




6. Structural abnormalities of the biliary tract can prevent drainage of bile from the canaliculus into the intestine and can cause accumulation of bile and reflux of bilirubin into the systemic circulation:
a. Biliary atresia (BA) is a progressive disease characterized by inflammation and fibrosis of the extrahepatic biliary tract resulting in partial or complete obliteration of the extrahepatic bile ducts.





b. Intrahepatic cholestasis is often associated with the histologic finding of bile duct paucity, defined as a reduced ratio of interlobular bile ducts to portal tracts (normal is 0.9 to 1.8; paucity is less than 0.5). Paucity of bile ducts may be syndromic (Alagille’s syndrome, which is associated with peripheral pulmonic stenosis, butterfly vertebrae, and characteristic facies). Many forms of intrahepatic cholestasis do not exhibit bile duct paucity. Treatment for all forms is symptomatic, with special consideration given to management of malnutrition and pruritus. Liver transplantation may be required in some cases.
 Progressive familial intrahepatic cholestasis, type I (PFIC-1), also known as Byler’s disease, is caused by defects in the FIC1 gene on chromosome 18q21–22. This P-type adenosine triphosphatase (ATPase) functions in transport of aminophospholipids across the hepatocyte canalicular plasma membrane. Patients characteristically have a low gamma glutamyltranspeptidase (GGTP) level. The average age of onset is 3 months, and progression to cirrhosis is variable. Watery diarrhea may occur, presumably secondary to intestinal absence of FIC1. Treatment is aimed at pruritus and poor growth. Liver transplantation is curative of the liver disease and typically is required in the first 2 decades of life. Benign recurrent intrahepatic cholestasis (BRIC) maps to the FIC1 locus, but the disease does not progress.
Progressive familial intrahepatic cholestasis, type I (PFIC-1), also known as Byler’s disease, is caused by defects in the FIC1 gene on chromosome 18q21–22. This P-type adenosine triphosphatase (ATPase) functions in transport of aminophospholipids across the hepatocyte canalicular plasma membrane. Patients characteristically have a low gamma glutamyltranspeptidase (GGTP) level. The average age of onset is 3 months, and progression to cirrhosis is variable. Watery diarrhea may occur, presumably secondary to intestinal absence of FIC1. Treatment is aimed at pruritus and poor growth. Liver transplantation is curative of the liver disease and typically is required in the first 2 decades of life. Benign recurrent intrahepatic cholestasis (BRIC) maps to the FIC1 locus, but the disease does not progress.
Progressive familial intrahepatic cholestasis, type I (PFIC-1), also known as Byler’s disease, is caused by defects in the FIC1 gene on chromosome 18q21–22. This P-type adenosine triphosphatase (ATPase) functions in transport of aminophospholipids across the hepatocyte canalicular plasma membrane. Patients characteristically have a low gamma glutamyltranspeptidase (GGTP) level. The average age of onset is 3 months, and progression to cirrhosis is variable. Watery diarrhea may occur, presumably secondary to intestinal absence of FIC1. Treatment is aimed at pruritus and poor growth. Liver transplantation is curative of the liver disease and typically is required in the first 2 decades of life. Benign recurrent intrahepatic cholestasis (BRIC) maps to the FIC1 locus, but the disease does not progress.





7. Alterations in the enterohepatic circulation can produce an increase in reabsorption of bilirubin from the intestine. The cause may be intestinal obstruction, as in intestinal atresia or Hirschsprung’s disease, or alterations in the bacterial flora by the use of antibiotics.
Complications
1. Unconjugated hyperbilirubinemia
 Kernicterus (bilirubin encephalopathy) may result from elevated levels of unconjugated bilirubin. Populations at risk include neonates and individuals with Crigler–Najjar syndrome, type I.
Kernicterus (bilirubin encephalopathy) may result from elevated levels of unconjugated bilirubin. Populations at risk include neonates and individuals with Crigler–Najjar syndrome, type I.




Kernicterus (bilirubin encephalopathy) may result from elevated levels of unconjugated bilirubin. Populations at risk include neonates and individuals with Crigler–Najjar syndrome, type I.

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
