ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Nonalcoholic fatty liver disease (NAFLD) is the term that represents a broad spectrum of disease ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) and steatofibrosis.
NAFLD is commonly associated with the metabolic syndrome, obesity, type 2 diabetes, and dyslipidemia; 80% of patients with the metabolic syndrome have NAFLD.
Patients generally present without clinical symptoms but with mild transaminase elevations; NAFLD is the most common cause of increased serum transaminase levels.
NAFLD is a clinical diagnosis after exclusion of other causes of liver disease.
Ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI) are useful for the detection.
Liver biopsy is currently required to distinguish NASH from NAFLD.
GENERAL CONSIDERATIONS
NAFLD is characterized by hepatic steatosis, the hepatocellular accumulation of triglycerides in the absence of significant alcohol consumption. Simple steatosis, or bland steatosis, connotes fat accumulation in the absence of hepatic inflammation. By contrast, NASH indicates the presence of inflammation and fibrosis in association with hepatic steatosis. NAFLD is sometimes used as an overarching term that includes simple steatosis and NASH, but can include NASH-related cirrhosis and advanced disease, or nonalcoholic steatofibrosis.
Whereas the histopathology of NAFLD and NASH is similar to that of alcohol-related liver disease, the etiology is quite distinct. Significant data from basic and clinical research have demonstrated that the metabolic underpinnings of NAFLD are rooted in insulin resistance. Indeed, NAFLD is commonly associated with other manifestations of insulin resistance including obesity, essential hypertension, type 2 diabetes mellitus, low levels of high-density lipoprotein (HDL), hypertriglyceridemia, and less commonly polycystic ovarian disease, or hypothyroidism. Although early studies suggested it to be a benign condition, it is now apparent that NAFLD has become a major cause of liver-related morbidity and mortality.
The absence of signs and symptoms, combined with a lack of sensitive and specific diagnostic tests, makes estimation of the prevalence of NAFLD difficult. Elevated liver enzymes are not sensitive for detecting NAFLD and there is no current consensus that histopathology is a gold standard for diagnosis. Although likely an underestimate for these reasons, the prevalence of NAFLD in the United States is considered to be in the range between 11% and 46% of the general population. The prevalence of NASH is significantly lower, in the range of 5–8%. As a result, the prevalences of NAFLD and NASH easily exceed chronic hepatitis C virus (HCV) infection, which afflicts 1.8% of the US population. A common polymorphism in the gene encoding patatin-like phospholipase-3 (PNPLA3) (synonym adiponutrin) is strongly associated with NAFLD (in some racial and ethnic groups) as well as histopathologic severity.
Population-based studies reveal that NAFLD is more common in men than women. It is more common in Hispanics compared with whites; and, more common in whites than blacks. It is assumed that the prevalence of NAFLD will increase over time in parallel with the growing epidemic of obesity and diabetes. Of particular concern is that NAFLD is increasing in the pediatric population, with prevalence estimated at around 3% of children and 20–50% of obese children.
[PubMed: 19818302]
[PubMed: 15565570]
[PubMed: 21700865]
[PubMed: 24222094]
[PubMed: 12512031]
Insulin resistance represents the most important risk factor for the development of NAFLD. Because insulin resistance is also the hallmark of the metabolic syndrome, it is not surprising that there is a close mechanistic link between NAFLD and the metabolic syndrome. The metabolic syndrome is generally defined as the coexistence of three or more of the following findings: (1) increased waist circumference, (2) hypertriglyceridemia, (3) hypertension, (4) elevated fasting plasma glucose, and (5) low HDL cholesterol level. Patients with metabolic syndrome have a 4- to 11-fold higher risk of developing NAFLD, and the prevalence of metabolic syndrome in patients with NAFLD ranges from 18% to 67%, depending on body weight. Patients who have NAFLD and metabolic syndrome sustain a threefold risk for NASH compared with NAFLD alone. It is possible that steatosis may simply characterize the hepatic manifestation of the metabolic syndrome. However, ethnic differences in NAFLD prevalence are still observed in patients with the metabolic syndrome or obesity, indicating that additional genetic factors play key roles.
There is also a close association of NAFLD with obesity. The prevalence of obesity in patients with NAFLD is reported to vary from 30% to 100%. In obese patients (body mass index [BMI] ≥30) the risk of NAFLD is elevated fivefold. Importantly, the frequency of NASH also varies in proportion to weight. The prevalence of NASH is 3% in the lean population, but rises to 19% in obesity and to nearly 50% in morbidly obese individuals. Consistent with the close relationship of NAFLD with the metabolic syndrome, NAFLD is more common in individuals with an abdominal concentration of fat, even at lower BMIs.
The prevalence of NAFLD is high in the type 2 diabetic population (50%), and the prevalence of type 2 diabetes in NAFLD patients ranges from 10% to 75%. The prevalence of NAFLD appears to increase as a continuous function of fasting plasma glucose. Importantly, NASH is disproportionately represented in type 2 diabetics, with significant fibrosis and cirrhosis present in approximately 20% of patients. In hyperlipidemic patients, the overall prevalence of fatty liver is 50%, with hypertriglyceridemia and mixed dyslipidemia conferring a fivefold increased risk of NAFLD. The prevalence of hyperlipidemia associated with NAFLD varies from 20% to 90%. In keeping with a strong link to the metabolic syndrome, low HDL cholesterol levels are also commonly observed in patients with NAFLD. Emerging evidence suggests that hypertension is linked to NAFLD through its relationship to insulin resistance.
[PubMed: 17311652]
[PubMed: 17498512]
[PubMed: 3683675]
[PubMed: 24613289]
PATHOGENESIS
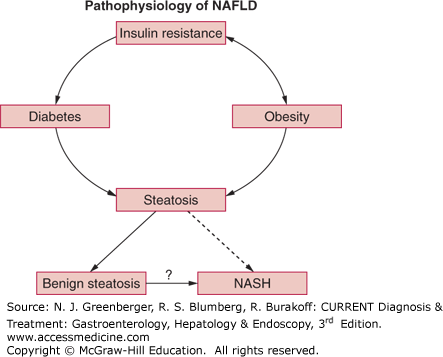
Current concepts indicate that insulin resistance is the primary metabolic defect leading to NAFLD (Figure 44–1). This leads to an influx into the liver of free (ie, nonesterified) fatty acids that are liberated from adipose tissues due to a failure of insulin to suppress hormone-sensitive lipase. In addition, elevated insulin and glucose levels associated with insulin resistance promote continued synthesis of triglycerides in the liver. These two sources of triglycerides result in lipid-engorged hepatocytes (ie, macrovesicular hepatic steatosis alone). The presence of NAFLD appears to be a relatively innocuous occurrence, and it does not appear that progressive liver injury necessarily ensues. Although many patients with NAFLD have coexisting type 2 diabetes, this is not uniformly true. Indeed, steatosis and insulin resistance may be present in the absence of other components of the metabolic syndrome.
Figure 44–1.
Insulin resistance is the foundation hepatic steatosis. Type 2 diabetes is a consequence of insulin resistance, but obesity can also result in insulin resistance. As indicated by the question mark, it is not known whether there is progression of benign steatosis to nonalcoholic steatohepatitis (NASH). NAFLD, nonalcoholic fatty liver disease. (Reproduced, with permission, from Parekh S, Anania FA. Abnormal lipid and glucose metabolism in obesity: implications for nonalcoholic fatty liver disease. Gastroenterology. 2007 May;132(6):2191–2207.)
A common, but not uniformly accepted hypothesis suggests that NASH evolves as a progression from NAFLD. In this regard, the presence of NALFD represents a first “hit.” According to this theory, NASH develops due to a second hit whereby hepatocytes laden with triglycerides are vulnerable to additional insult. Although definitive data to support individual second hits are lacking, two categories have been described and have gained some acceptance. These are oxidative stress and specific cytokines plus lipopolysaccharides. Free fatty acids and hyperinsulinemia potentiate lipid peroxidation and the release of hydroxy free radicals, which directly injure hepatocytes by recruitment of necroinflammatory mediators. Chronic liver injury sustained over time will then lead to activation of hepatic stellate cells, creating the potential for hepatic fibrosis.
At least in certain animal models of NASH, lipopolysaccharides are released into the portal vein and are potent agonists for tumor necrosis factor-α (TNF-α) production, with the subsequent release of interleukins. Interestingly, the same mechanisms appear to be pathogenic in alcoholic hepatitis and produce quite similar histopathologic lesions. The combination of oxidative stress and necroinflammatory cytokines is associated with compromised mitochondrial function, which impairs electron transport and depletes adenosine triphosphate. This renders hepatocytes incapable of handling the enhanced oxidative stress. In this setting, the increased oxidative stress may overwhelm the intrinsic antioxidant capacity of the liver. Intracellular free fatty acids within the liver also promote endoplasmic reticulum stress and mitochondrial damage, which lead to increase lipid synthesis and hepatocyte apoptosis via activation of c-Jun N-terminal kinase (JNK).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


