According to this model, the transition from endocrine to paracrine-driven prostate cancer is associated with a number of modifications involving both oncogenes activation and tumor suppressor genes loss (e.g., loss of the PTEN tumor suppressor protein leads to uncontrolled activity of the phosphoinositide 3-kinase (PI3K)/Akt pathway). During the microenvironment-dependent phase, cancer progression is dominated by tumor adaptive changes and these modifications will end up in a vicious circle, in which the microenvironment alters the tumors and the tumor in turn modify the microenvironment. Emerging evidences suggest that there is a bidirectional crosstalk between several oncogenes and AR; therefore, activation of oncogenes and androgen depletion are not independent functions [8]. Among main changes we can identify the following:
Upregulation of enzymes involved in steroidogenesis—particularly intratumoral steroidogenesis and in the adrenal glands.
AR gene amplifications that lead to an AR overexpression and therefore possibly a hypersensitivity to androgen ligands. Castration-resistant patients with AR amplification respond at a higher rate to second-line maximal androgen blockade when compared to those without amplification [10].
Although castration, the hypersensitivity of AR to low levels of androgens results in a continuous tumor growth.
Prostate cancer (PC) xenograft models demonstrate how an increased expression of AR is the main aberration found in ADT refractory tumors and a high level of AR is overexpressed in CRPC tumors [2].
AR mutations: the AR gene is the most mutated type of steroid receptor. Most of the AR mutations are gain-of-function mutations. These can be mapped on the ligand-binding domain and eventually will result into androgen hypersensitivity or AR decreased ligand specificity. More than 660 AR mutations have been reported thus far [11]. The presence of mutation of AR in Caucasians with an untreated localized PCa is <2 %, but the frequency of AR mutations may vary ranging between 20 and 50 % in metastatic castration-resistant tumors. These mutations may involve false triggering from other steroid hormones. These mutations allow AR to be activated by noncanonical ligands, like other steroids [2] or from treatments initially used as antiandrogens. The latter phenomenon is known as “antagonist-to-agonist” conversion and explains the “antiandrogen withdrawal” effect, i.e., the decline in PSA manifested in ~25 % of patients following the discontinuation of these drugs after clinical progression [1, 12, 13].
Epigenetic modifications such as methylation of the AR receptor and AR splicing variant in promoting a constitutive gene expression. The role of co-activators and co-repressors in the development of castration-resistant disease is still not completely clear and so is it the role of AR post-translational modifications (PTMs). Most of the PTMs have not been studied within the in vivo human context and they will need to be translated into clinical experience to understand clinical utility [14].
10.2.2 Clinical Experience
10.2.2.1 Role of Taxanes in CRPC
Docetaxel-based chemotherapy is the established standard of care at development of castration-resistant prostate cancer. This drug was proven to be superior to the previous standard of care represented by mitoxantrone. The latter was assessed in a randomized phase III trial in which patients were randomized to mitoxantrone and corticosteroids vs. single use of steroids. Mitoxantrone in improvement of pain, QoL, and PSA response, but overall survival (OS) was similar [15, 16]. The first trial showing superiority of docetaxel over mitoxantrone was the SWOG 99-16, in which the combination of docetaxel and estramustine was demonstrated to be superior to mitoxantrone and prednisone [17, 18]. However, the main phase III trial which led to registration of docetaxel as standard of care for progressing CRPC was the TAX327; in this trial, 1,006 men with metastatic CRPC received 5 mg of prednisone twice daily and were randomly assigned to receive ten courses of 3-weekly mitoxantrone or weekly (5 weeks out of 6) docetaxel or 3-weekly docetaxel, respectively. The study proved that 3-weekly docetaxel and prednisone was the preferred treatment leading to an improvement of overall survival and quality of life with a better pain control and a better biochemical response [19]. According to this trial, docetaxel given every 3 weeks has become the standard of care in symptomatic CPRC patients.
Apart from a direct cytotoxic activity, there is increasing interest and at least in vitro evidence of an “antihormonal” mechanism of action of docetaxel and taxanes in general [20]. Decreased expression of AR-transactivated genes through the inhibition of AR transcriptional activity represents a new potential molecular pathway that could explain efficacy of taxanes in the treatment of prostate cancer [21].
Cabazitaxel (JEVTANA®)
Cabazitaxel was the first demonstrated drug to improve survival in patients with CRPC progressing after treatment with docetaxel. This drug is a member of the taxane family, which includes paclitaxel and docetaxel. Cabazitaxel binds to tubulin and promotes assembly into microtubules and inhibits disassembly, resulting in stabilization of microtubules, which interferes with mitotic and cellular interphase activity [22]. It shows a power comparable to that of docetaxel in cellular line but the uniqueness of having antitumor activity in models resistant to paclitaxel and docetaxel [23–25], and also the ability to cross the blood–brain barrier.
The study that led to the approval by the FDA of cabazitaxel was the TROPIC, an open-label randomized phase III trial in men with metastatic castration-resistant prostate cancer who had received previous hormone therapy, but whose disease had progressed during or after treatment with a docetaxel-containing regimen.
In this study, 755 patients were enrolled, 378 received cabazitaxel 25 mg/m2 intravenously over 1 h and oral prednisone 10 mg daily. Patients under treatment with LHRH agonists and bisphosphonates continued treatment along the study.
Eligible patients were aged at least 18 years, with an Eastern Cooperative Oncology Group (ECOG) PS 0–2. Patients who had previous mitoxantrone therapy, radiotherapy to 40 % or more of the bone marrow, or cancer therapy (other LHRH analogues) within 4 weeks before enrollment were excluded. Patients with measurable disease were required to have disease progression with at least one visceral or soft-tissue metastatic lesion. Patients with nonmeasurable disease were required to have rising PSA in two consecutive measurements or at least one new demonstrable radiographic lesion before enrollment; adequate hematological, hepatic, renal, and cardiac function, and a left-ventricular ejection fraction ≥50 % assessed by multi-gated radionuclide angiography or echocardiogram.
Results proved that median survival in people taking cabazitaxel was 15.1 months vs. 12.7 months in the mitoxantrone group. The hazard ratio for death of men treated with cabazitaxel compared with those taking mitoxantrone was 0.70 (95 % CI 0.59–0.83, P < 0.0001). The median progression-free survival was 2.8 months compared with 1.4 of mitoxantrone (HR 0.74, 0.64–0.86, P < 0.0001). Most significant adverse were neutropenia (cabazitaxel vs. mitoxantrone 83 58 %), diarrhea (6 % vs. 1 %), and febrile neutropenia (8 % vs. 1 %). The high incidence of febrile neutropenia suggests that treatment requires prophylaxis [26].
An update of the study to 25.5 months also shows more patients remained alive following cabazitaxel than mitoxantrone [odds ratio 2.11; 95 % confidence interval (CI) 1.33–3.33]. Treatment with cabazitaxel was prognostic for survival ≥2 years; in fact, the probability of surviving ≥2 years was 27 % (95 % CI 23–32 %) with cabazitaxel vs. 16 % (95 % CI 12–20 %) with mitoxantrone. Pain at baseline and pain response were comparable between treatment groups. Average daily pain performance index was lower for cabazitaxel vs. mitoxantrone (all cycles; 95 % CI −0.27 to −0.01; P = 0.035) and analgesic scores were similar. Grade ≥3 peripheral neuropathies were uncommon and comparable between treatment groups [27].
A phase III clinical trial comparing the efficacy and toxicity of cabazitaxel 20 mg/m2 prednisone vs. cabazitaxel 25 mg/m2 (PROSELICA; NCT01308580) is ongoing [28].
Cabazitaxel is also being evaluated in a first-line phase III trial (FIRSTANA; NCT01308567). In this open-label trial, 1,170 patients with chemotherapy-naïve mCRPC will be randomized to docetaxel (75 mg/m2 every 3 weeks) or cabazitaxel (20 or 25 mg/m2 every 3 weeks) plus prednisone (10 mg orally, daily) [29].
10.2.2.2 CYP17 Inhibitors
Abiraterone (Zytiga®)
Abiraterone acetate is new potent hormonotherapy recently developed and approved for treatment of CRPC. This drug represents the “proof-of-concept” that prostate cancer remains hormone driven even after failure of androgen deprivation therapy. The reason of developing such a drug started off from the observation that patients exposed to ketoconazole—a known antifungal treatment essentially able to inhibit the CYP17 enzyme which is key in male sex hormones production—could achieve some response at least in terms of PSA drop because of its ability to prevent prostate cancer cells growth. However, ketoconazole is poorly tolerated and its affinity for CYP17 is low. Research conducted at the Institute of cancer Research UK led to the development of a potent CYP17 inhibitor, which was called CB7598 or abiraterone. However, this drug raised the question of the potential-related side effects, namely the risk of causing adrenal insufficiency. However, the clinical observation that children born with a deficiency of the CYP17 enzyme did not develop adrenal insufficiency (http://www.icr.ac.uk/press/recent_featured_articles/Story_Abiraterone/index.shtml), led to the initial clinical application of the drug (Fig. 10.2).
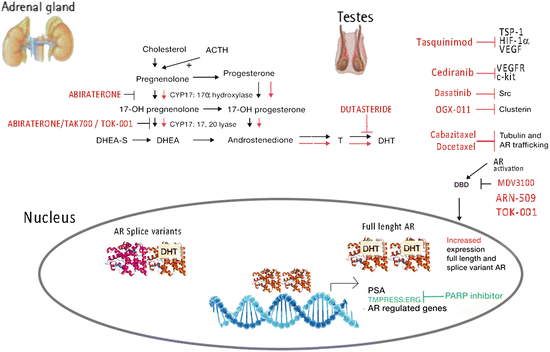
Fig. 10.2
Mechanism of action of new drugs in the treatment of CRPC
The phase I and II trials carried out in men with castration-resistant prostate cancer showed that the drug was very well tolerated with no major side effects and also some interesting preliminary efficacy in terms substantial PSA decline. Radiological response was also reported with evidence of partial responses in patients with measurable disease [30]. The phase III international, randomized, double-blind, placebo-controlled trial called COU-AA-301 finally led to the approval of abiraterone acetate in castration-resistant prostate cancer patients previously exposed to docetaxel. In this trial, 1,195 patients received prednisolone and 1 g of abiraterone acetate or placebo on a fasted status daily. Treatment could be continued until PSA, radiological and clinical disease progression was documented. Results of the final results analysis—updated in 2012—show that group taking abiraterone of 15.8 months compared to 11.2 months in the group taking placebo (hazard ratio [HR] 0.74, 95 % CI 0.64–0.86, P < 0.0001). Median time to PSA progression was 8.5 months for the group treated with abiraterone vs. 6.6 months (HR 0.63, 0.52–0.78, P < 0.0001), similar to the median radiologic progression-free survival (5.6 months vs. 3.6 months, HR 0.66, 12:58–0.76, P < 0.0001). PSA responses (29.5 % vs. 5.5 %, P < 0.0001) were all improved in the abiraterone group compared with placebo group. Adverse events grade 3–4 related to the use of abiraterone were: fatigue, anemia, back pain, and bone pain [31]. The approval from the U.S. Food and Drug Administration (FDA) for hormone refractory prostate cancer, in post-docetaxel setting, was given in April 2011 and the European approval was followed in September 2011. More recently, abiraterone has been investigated in asymptomatic or paucisymptomatic patients with CRPC before administration of docetaxel. Based on experiences of previous earlier phase of development trials [32–34], investigated the potential use of abiraterone acetate in 1,088 castration-resistant patients not previously treated with docetaxel were randomized to receive prednisone and abiraterone or placebo in phase III international, double-blind, placebo-controlled study. The results from an interim analysis showed that the median radiographic progression-free survival (PFS) was 16.5 months with the use of abiraterone–prednisone and 8.3 months with only prednisone (HR, 0.53; 95 % [CI], 0.45–0.62, P < 0.001). However, on a follow-up of 22.2 months, the primary end point of the trial overall survival (OS) was not met as there were not enough related events in the abiraterone arm (vs. 27.2 months for prednisone alone, HR 0.75, 95 % CI, 0.61–0.93, P = 0.01). The drug also demonstrated superiority over placebo, for opiate use for cancer-related pain, prostate-specific antigen progression, and decline in performance status [35]. Nevertheless, based on these results abiraterone acetate was approved by FDA in the predocetaxel setting in December 2012. In keeping with its clinical efficacy, abiraterone is able to affect the detection of circulating tumor cells with the Cell Search machine and results seem to correlate closely with overall survival of patients. Studies are ongoing to assess the efficacy of abiraterone in combination with LHRH analogues is hormonotherapy naïve patients (NCT01088529 and NCT00924469) as well as pre and concomitant to radical radiotherapy (NCT01023061). Eventually, patients exposed to abiraterone acetate will develop resistance to this therapy; therefore, the basic and clinical research is aiming to understand mechanism and develop strategies to overcome that. Combination treatments with chemotherapy [36] or other agents, such as PI3K (phosphoinositide 3-kinase)/AKT (a serine/threonine protein kinase) inhibitors and PARP inhibitors [37], are ongoing and results from clinical experience are awaited [38].
TOK-001 (Galeterone)
Galeterone (also known as TOK-001) was rationally designed to inhibit the human CYP17 enzyme, but was also found to be a potent pure AR antagonist and to effectively prevent the binding of synthetic androgens to both mutant and wild-type AR [39, 40] (Fig. 10.2). A phase I, multicentre, dose-finding study in patients with chemotherapy naïve CRPC was published in the American Society of Clinical Oncology (ASCO) meeting in 2012. Patients were enrolled in cohorts from 650 to 2,600 mg of TOK-001 daily, with 36 of 49 completing the 12-week treatment course. The treatment was generally well tolerated, with only one severe AE considered related to TOK-001 (rhabdomyolysis with acute renal failure, with high-dose statin use). Overall 22 % of treated patients had a >50 % PSA level decline, and an additional 26 % had 30–50 % PSA level declines. Consistent with lyase inhibition, increased corticosteroids, and suppressed androgens were seen with dose escalation [41].
Tak-700 (Ortonel)
TAK-700 selectively inhibits the CYP17A 17, 20 lyase [42], whose task is to reduce in vivo levels of androgens produced by the adrenal gland (Fig. 10.2). Dreicer et al, tested tak-700 in a phase I/II open-label, dose-escalation to test safety in 26 mCRPC patients of oral twice daily (BID) TAK-700. All patients treated with ≥300 mg had a PSA decrease, of 15 patients who received TAK-700 ≥ 300 mg for ≥3 cycles and had a 3-month PSA determination, 12 (80 %) had ≥50 % PSA reductions and 4 (27 %) had ≥90 % reductions. Median testosterone and DHEA sulfate levels decreased from 5.5 to 0.6 ng/dL and from 50.0 μg/dL [43].
10.2.2.3 AR Antagonists
Enzalutamide (Xtandi®)
Enzalutamide (formerly MDV3100) is an androgen receptor signaling inhibitor chosen for clinical development on the basis of activity in prostate-cancer models with overexpression of the androgen receptor. Enzalutamide is distinct from the currently available antiandrogen agents that inhibit nuclear translocation of androgen receptor, DNA binding, and coactivator recruitment. It also has a greater affinity for the receptor, induces tumor shrinkage in xenograft models (conventional agents only retard growth), and has no known agonistic effects [13, 44] (Fig. 10.2).
Enzalutamide has been approved by the FDA in July 2012 for CRPC patients, following docetaxel chemotherapy.
Enzalutamide is administered once daily, without the need for concomitant prednisone which has been postulated to activate androgen receptor signaling.
One of the first studies of Scher’s team showed that in a population of 140 subjects there was at least a PSA halving in 56 % of patients, soft tissue response in 22 %, stable bone disease in 56 %, and a reduction below threshold levels of circulating tumor cells in 49 %; moreover, there was a median time to radiological progression of 47 weeks. Among adverse effects of grade 3–4 11 % complained fatigue usually resolved with dose reduction. So results proved that MDV3100 has a substantial antitumor activity in men with castration-resistant prostate cancer, whether or not they have been exposed to chemotherapy [5].
The potential of the drug has been confirmed by analysis of the AFFIRM group (A Study Evaluating the Efficacy and Safety of the Investigational Drug MDV3100) that enlists 1,199 subjects in a phase III, double-blind, placebo-controlled trial. Among inclusion criteria of the study were presence of testosterone levels <50 ng/dL [1.7 nmol/L], previous treatment with docetaxel and progressive disease defined according to Functional PCWG2 criteria. The use of prednisone or other glucocorticoids was allowed but not required. Important to note that patients were maintained on GnRH agonist/antagonist therapy and could receive supportive care medications.
The median overall survival was 18.4 months in the group treated with the enzalutamide respect to 13.6 months in the placebo group (hazard ratio for death in the enzalutamide group, 0.63, 95 % CI, 0.75–0.53, P < 0.001). The enzalutamide declared itself superior over placebo in all secondary end points: the proportion of patients with a reduction in the prostate-specific antigen (PSA) level by 50 % or more (54 % vs. 2 %, P < 0.001), the soft-tissue response rate (29 % vs. 4 %, P < 0.001), the quality-of-life response rate (43 % vs. 18 %, P < 0.001), the time to PSA progression (8.3 vs. 3.0 months, hazard ratio, 0.25, P < 0.001), radiographic progression-free survival (8.3 vs. 2.9 months, hazard ratio, 0.40; P < 0.001), and the time to the first skeletal-related event (16.7 vs. 13.3 months, hazard ratio 0.69, P < 0.001).
Among the major adverse events related to the use of MDV3100 stands fatigue, lightheadedness, and hot flushes. It is appropriate to specify how the average time from onset of any adverse event of grade 3 or higher was 8.4 months longer in the group that took enzalutamide than placebo group (12.6 vs. 4.2 months), so leading to an improvement in long-term control of disease without an increase of the reactions events.
Should also be noted that in 0.6 % of the population examined occurred seizures, in some cases in people with predisposing conditions or concomitant treatments: physicians should pay particular attention when administering enzalutamide in patients with a history of epilepsy or with predisposing factors (underlying brain injury, stroke, brain metastases, alcoholism, or patients taking drugs that lower the seizure threshold) [18].
In order to better understand the potential use of the drug is now ongoing a phase III trial in chemo-naïve patients (PREVAIL; NCT01212991).
ARN-509
ARN-509 is a drug able to inhibit AR nuclear translocation, AR binding to androgen response elements and, unlike bicalutamide, does not exhibit agonist properties in the context of AR overexpression (Fig. 10.2). In its first inhuman phase I study, PSA declines at 12 weeks (≥50 % reduction from baseline) were observed in 46.7 % of patients. Reduction in FDHT uptake was present at all doses, with a plateau in response at ≥120-mg dose, consistent with saturation of AR binding. Assessed in this study were also safety, tolerability, pharmacokinetics, pharmacodynamics, and antitumor activity of ARN-509 in men with metastatic CRPC [45].
10.2.2.4 Immunotherapy
Sipuleucel-T (Provenge®)
Immunotherapy is emerging as a therapeutic strategy for patients with prostate cancer. There are numerous strategies approachable in the use of the immune system and between them stands the production of cancer vaccines designed to stimulate the immune cells against target expressed by cancer cells.
Sipuleucel-T (APC8015) is an autologous active cellular immunotherapy used in the treatment of men with asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer (CRPC). It is the first therapeutic cancer vaccine to receive U.S. FDA approval. Sipuleucel-T is an “active cellular immunotherapy,” a type of cancer vaccine consisting of autologous peripheral blood mononuclear cells (PBMCs), including antigen-presenting cells (APCs), which were activated ex vivo with a recombinant fusion protein (PA2024). The APCs are then incubated with a recombinant protein composed of prostatic acid phosphatase (PAP) linked to granulocyte-macrophage colony-stimulating factor (GM-CSF) [46]. PAP was chosen as the target antigen because is expressed in prostate tissues and in the vast majority of carcinomas of the prostate with a no or minimal expression in other tissues [47], also does not share a high degree of sequence homology with any other protein know. Task of GM-CSF is instead to increase the uptake of APCs [48].
Approximately 3 days prior to each infusion of sipuleucel-T, patients undergo a leukapheresis procedure for collection of autologous peripheral blood mononuclear cells. Preparation of sipuleucel-T Involves enrichment for antigen-presenting cells from the leukapheresis product and activation ex vivo with a recombinant fusion protein (PA2024).
As early as 2006, the randomized, placebo-controlled study of Small and colleagues showed how, by analyzing 127 patients, the median time to disease progression (TTP) for sipuleucel-T was 11.7 weeks compared with 10.0 weeks for placebo (P = 0.052, log-rank; hazard ratio [HR], 1.45; 95 % CI, 0.99–2.11). Survival rate at 3 years was stated to 34.1 %, with an average increase of 4.5 months and a median survival of 25.9 months. The treatment was also well tolerated by patients. It is important to note, however, as the primary end point (TTP) has not been reached [49].
A second study of 225 patients from the group of Higano showed a trend of increase in survival with sipuleucel-T despite not statistically significant. There was also an insignificant effect on time to disease progression (PFS) [50].
Fundamental to approve the sipuleucel-T was the phase III study called Immunotherapy for Prostate adenocarcinoma Treatment (IMPACT). Structured as a double-blind, placebo-controlled, multicenter trial enrolled patients with any Gleason score, asymptomatic or minimally symptomatic, PSA ≥ 5 ng/mL, serum testosterone < 50 ng/dL (17 nmol/L). Exclusion criteria included a baseline ECOG PS greater ≥2, presence of visceral metastases, pathological fractures of long bones, compression of the spinal cord, within 28 days of treatment with glucocorticoids, prior external beam radiation, surgery, or systemic treatment for cancer prostate (except for medical or surgical castration). Exclusion criteria include also the use of bisphosphonates in previous 28 days, history of treatment with two or more chemotherapy regimens, or chemotherapy within 3 months. The continuation of chemical castration or bisphosphonate therapy has been requested at least until the time of disease progression.
All 512 patients were randomly assigned in a 2:1 ratio to receive sipuleucel-T or placebo every 2 weeks for three infusions. Subjects enrolled had received prior androgen deprivation. Be mentioned that the average age was 71 years.
Results of the analysis proved as in the group with sipuleucel-T the adjusted hazard ratio for death was 0.78 (95 % CI, 0.61–0.98) and that reduction in the risk of death was 22 % (P = 0.03). The median survival of 4.1 months stood higher than the control group (25.8 months vs. 21.7 months). The probability of survival at 36 months was 31.7 % in the group taking sipuleucel-T and 23.0 % in the placebo group.
The median time to progression disease was 14.6 weeks (3.7 months) in the group treated with sipuleucel-T and 14.4 weeks (3.6 months) in the placebo group (hazard ratio 0.92, 95 % CI, 0.75–1.12, P = 0:40). Among patients with periodic evaluations of PSA, a reduction of at least 50 % was observed in 8 of 311 patients (2.6 %) in subjects treated with sipuleucel-T compared to 1.3 % of the control group (2 of 152 patients).
Among the major adverse effects experienced in the group taking sipuleucel-T was found were chills, fever (pyrexia), headache, flu-like illness, myalgia, hypertension, hyperhidrosis, and groin pain. Except for groin pain, most of these events have occurred the day following the infusion and resolved within 1 or 2 days. Overall, only 3 of 338 patients (0.9 %) in the group sipuleucel-T were not able to receive all three infusions because of adverse events related to the infusion [51].
Due to these results in April 2010, the Food and Drug Administration (FDA) has authorized sipuleucel-T in the treatment of metastatic castration-resistant disease in patients asymptomatic or minimally symptomatic.
Although there are no short-term changes in disease progression or available biomarkers to assess response, these agents appear to improve survival. One hypothesis suggests that this apparent paradox can be explained by the growth-moderating effects of treatments, which do not cause tumor size to diminish, but rather stall or slow their growth rate over time [52]. For this reason, further studies should clarify the real benefits in relation to other treatments.
PROSTVAC (TRICOM®)
In development is also PROSTVAC a fowlpox vaccines and vectors with a triade of costimulatory molecule transgenes, including intercellular adhesion molecule 1 (ICAM-1), B7.1, and leukocyte function-associated antigen 3 (LFA-3), which has been designated TRICOM® [53]. In a randomized double-blind phase II trial, patients with a mild or asymptomatic metastatic chemo-naive PCa, Gleason score ≤7 and rising PSA were randomized to receive placebo or PROSTVAC. Patients on the vaccine arm achieved an improved median survival of 24.5 months compared with 16 months in the placebo group P = 0.016 [54]. T-cell activation is dependent on two separate signals: the first is the presentation of an antigen linked to the major histocompatibility complex (MHC) molecule and the second is a costimulatory of the interaction of CD28 on the T cell with CD80 and CD86 on the APC. Both CD28 and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) are expressed on T cells, and competitively bind to the same ligands present on APCs. In contrast to the activating function of CD28 however, CTLA-4 serves as an immunomodulatory molecule that negatively regulates T-cell activation and dampens the immune response generated. This inhibitory feedback is integral for the maintenance of peripheral tolerance of self-antigens and can be exploited therapeutically by targeting CTLA-4 to enhance T-cell-mediated antitumor immune responses.
Similarly, PROVASTAC VF showed a statistically significant survival difference of 8.5 months (25.1 vs. 16.6 months, estimated 0.56 HR 95 % CI, 0.85–12:37; stratified log-rank P = 0.0061) in a randomized, placebo-controlled trial [55]. Another single-arm phase II trial of PROSTVAC in mCRPC patients proved similar survival outcomes, with a median OS of 26.6 months [56]. A phase II trial randomized 26 patients with nonmetastatic CRPC to receive flutamide alone or with PROSTVAC-V/F reported preliminary that patients in the combination arm had improved time to progression compared with patients who received flutamide alone (223 vs. 85 days, respectively) [57].
Ipilimumab (Yervoy®)
Ipilimumab is a humanized monoclonal antibody against cytotoxic T lymphocyte antigen-4 (CTLA-4). CTLA-4, a T-cell surface receptor, is a key negative regulator of T-cell responses. Thus, ipilimumab augments antitumor immune response by inhibiting a negative regulator of T cells. Showing useful in some phase II studies with both castration [58] and PSA-TRICOM [59], serious difficulties were in the phase III trial CA-184-043 trial (Study 043) that enrolled 799 patients with mCRPC who had received prior docetaxel. In this trial, patients were randomized in a 1:1 ratio to receive bone-directed radiation therapy at 8 Gy followed by ipilimumab at 10 mg/kg (n = 399) or placebo (n = 400). A subgroup analysis suggests that patients with less advanced disease could still benefit from treatment with ipilimumab. The study failed to meet its primary end point of prolongation in overall survival (OS). Despite the lack of an OS benefit, progression-free survival (PFS) and a marked reduction in PSA were observed with immunotherapy.
Also, interesting is the trial by Madan et al. concerning ipilimumab in combination with a poxviral-based vaccine targeting prostate-specific antigen (PSA) and containing transgenes for T-cell costimulatory molecule expression, including CD80. Only one of the six patients previously treated with chemotherapy had a PSA decline from baseline. Of the 24 patients who were chemotherapy-naive, 14 (58 %) had PSA declines from baseline, of which six were greater than 50 %. The use of a vaccine targeting PSA that also enhances costimulation of the immune system did not seem to exacerbate the immune-related adverse events associated with ipilimumab [59]. Randomized trials are needed to further assess clinical outcomes of the combination of ipilimumab and vaccine in mCRPC.
10.2.2.5 Antiangiogenic Agents
Tasquinimod
Treatment with tasquinimod (TASQ) plays a role in the upregulation of TSP-1 expression and downregulation of HIF-1α and VEGF [60, 61] (Fig. 10.2). Preclinical data demonstrated that work synergistically with taxanes (docetaxel and cabazitaxel) [62, 63].
In a phase II trial by Pili et al., 201 patients were evaluated (134 assigned to TASQ; 67 to placebo). Six-month progression-free proportions for TASQ and placebo groups were 69 % and 37 %, respectively (P < 0.001), and median progression-free survival (PFS) was 7.6 vs. 3.3 months (P = 0.0042). So, TASQ significantly slowed progression and improved PFS in patients with metastatic CRPC with an acceptable AE profile [63]. Definitive assessment of tasquinimod in CRPC clinical treatment is still required and results of an ongoing major phase III clinical trials are awaited.
10.2.2.6 Tyrosine Kinase Inhibitors
Dasatinib (Sprycel®)
Dasatinib is an inhibitor of numerous kinases: BCR-ABL, SRC family (SRC, LCK, YES, and FYN), c-KIT, EphA2, and PDGFRβ. It is involved in multiple signaling pathways in prostate cancer and promotes tumor cell proliferation, survival, migration, and the transition to androgen independence. In experiments, dasatinib seems able to reduce the proliferation of osteoclasts and the release of calcium [64] (Fig. 10.2).
Related posts:
 Early Diagnosis of Failure After Primary Treatment: Multiparametric MRI Versus PET-TC
Early Diagnosis of Failure After Primary Treatment: Multiparametric MRI Versus PET-TC
 Prostate Cancer Units: How and Why
Prostate Cancer Units: How and Why
 Comparison Between a Multidisciplinary and a Monodisciplinary Approach to Prostate Cancer: Our 1-Year Experience
Intermittent Androgen Deprivation in the New Era: The Role of Urologist and Oncologist in a Multidisciplinary Team (MDT)
Comparison Between a Multidisciplinary and a Monodisciplinary Approach to Prostate Cancer: Our 1-Year Experience
Intermittent Androgen Deprivation in the New Era: The Role of Urologist and Oncologist in a Multidisciplinary Team (MDT)
 Modern Imaging in the Initial Diagnosis: The Role of the Radiologist in an MDT
Modern Imaging in the Initial Diagnosis: The Role of the Radiologist in an MDT
 Role of Pathology in the Multidisciplinary Management of Patients with Prostate Cancer
Role of Pathology in the Multidisciplinary Management of Patients with Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


