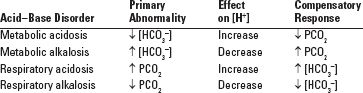
■ Acid–base homeostasis is dependent on the following reactions: [H+] + [HCO3−] ← → H2CO3 ← → H2O + CO2 (excreted by lungs) A primary change in either [HCO3−] or PCO2 will produce an adaptive (compensatory) change in the other in the same direction, in an attempt to keep pH constant (Tables 7.1 and 7.2) (Javaheri et al., 1982; Madias, 2010; Pierce et al., 1970).
TABLE | The Four Primary Acid Base Disorders and Their Compensatory Responses |
7.1 |
|
Renal Regulation of Acid–Base Balance
■ Reabsorption of filtered bicarbonate. Most or all of bicarbonate filtered at the glomerulus is reabsorbed, primarily in the proximal tubule. Failure of this reabsorption mechanism leads to proximal renal tubular acidosis (RTA).
■ Excretion of acid. The kidney excretes acid in three fashions:
■ Free hydrogen ion (H+) excretion (lowers urine pH)—this is quantitatively the least important mechanism
■ Titratable acid excretion (secreted H+ combines with poorly reabsorbable anions such as phosphate)
■ Excretion of ammonium ion (NH4+). The kidney generates ammonia (NH3) which combines with secreted H+ and is excreted as ammonium ion.
Failure of H+ secretion and/or ammonia production/ammonium excretion result in distal RTA.
■ High anion gap (AG) acidosis
The AG is a concept to give a clue as to the cause of metabolic acidosis (Gabow et al., 1980). Electroneutrality demands that the number of positive charges equals the number of negative charges (ions) in body fluids. Therefore, in plasma
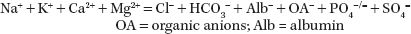
Simplifying this equation to include only certain monovalent measured ions and calling the other ions unmeasured cations (UC) or unmeasured anions (UA):
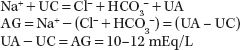
Overproduction of an endogenous organic acid (H+A−), where A− is the acid anion, leads to high-AG metabolic acidosis (e.g., with lactic acidosis A− = lactate). When lactic acid is produced, the H+ is titrated by bicarbonate as follows:
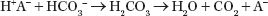
This reaction consumes 1 mol of bicarbonate for each mole of acid produced. If A− is not excreted or metabolized, it will accumulate in plasma, resulting in an AG. Theoretically, with this type of acidosis, the increase in AG will equal the decrease in bicarbonate concentration (see Supplement for examples).
■ Normal-AG (hyperchloremic) acidosis
Metabolic acidosis can also occur due to decreased renal acid excretion leading to retention of acid (H+ ions), which titrate extracellular bicarbonate, or it can be caused by loss of bicarbonate from the body. In either instance, the decrease in filtered bicarbonate results in decreased renal bicarbonate and increased renal chloride reabsorption (to maintain electoneutrality), resulting in a hyperchloremic acidosis. The plasma chloride rises to the same extent as the plasma bicarbonate falls, and the anion gap (AG) remains normal.
Causes of Metabolic Acidosis
■ High-AG acidosis
■ Increased organic acid production (with organic anion shown in parenthesis)
• Lactic acidosis (lactate)
• Ketoacidosis (acetoacetate, ß-hydroxybutyrate)
• Toxin ingestion
 Salicylate (mostly lactate)
Salicylate (mostly lactate)
 Methanol (formate)
Methanol (formate)
 Ethylene glycol (glyoxylate, oxalate)
Ethylene glycol (glyoxylate, oxalate)
■ Failure to excrete inorganic anions (phosphate, sulfate) in renal failure
■ Normal-AG (hyperchloremic) acidosis
■ Gastrointestinal loss of bicarbonate (diarrhea)
■ Renal loss of bicarbonate
• Proximal RTA (type 2) (Morris, 1969) (see also Chapter 2)
• Carbonic anhydrase (CA) inhibitors (prevent proximal HCO3− reabsorption)
■ Failure to excrete acid
• Distal RTA (type 1, type 4) (Morris, 1969) (see also Chapter 2)
• Renal failure
■ Administration of acid
• Infusion of HCl or its congeners (e.g., ammonium chloride)
■ Administration of large amounts of saline (dilutional acidosis)
■ Ureteral diversion (increased chloride–bicarbonate exchange resulting in bicarbonaturia and/or increased ammonium uptake, which is converted to ammonia and H+ in the liver; generally, only seen if there is partial obstruction of the conduit)
Treatment of Metabolic Acidosis
■ Treat underlying cause(s)
■ Bicarbonate (especially with normal-AG acidosis)—Na or K bicarbonate (or bicarbonate former) depending on etiology and electrolyte values
METABOLIC ALKALOSIS
Metabolic alkalosis is defined as a primary increase in the bicarbonate concentration, that is, [HCO3−] of the plasma. In primary metabolic alkalosis, the pH of the blood will be elevated (alkalemia). An increase in [HCO3−] of 1.0 mEq per L should be accompanied by an increase in PCO2 of 0.7 mm Hg (this is termed “respiratory adaptation”). The rise in PCO2 will keep the blood pH near normal (although mild alkalemia will be present). Metabolic alkalosis is also associated with a slightly elevated AG (Madias et al., 1979).
Generation of Metabolic Alkalosis
There are three ways to generate metabolic alkalosis:
■ Net loss of hydrogen ions (H+) from extracellular fluid (ECF)
■ Causes of H+ loss
• Gastrointestinal (GI)—loss of HCl from stomach (vomiting, gastric drainage)
HCl secretion by parietal cells of stomach does not normally cause metabolic alkalosis because HCl is titrated by pancreatic sodium bicarbonate:
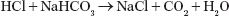
However, if HCl is lost from the body because of vomiting or gastric drainage, there is a net gain of bicarbonate. Metabolic alkalosis is then maintained by increased renal bicarbonate reabsorption due to depletion of ECF and chloride (see section Maintenance of Metabolic Alkalosis below).
• Renal—loss of H+ into urine (mineralocorticoid excess states) Hydrogen ions generated by protein metabolism are normally excreted by the kidneys primarily in the form of ammonium (NH4+) ions. In certain conditions, an inappropriate increase in renal H+ excretion leads to metabolic alkalosis. This is characteristic of primary mineralocorticoid excess states. Aldosterone (the naturally occurring mineralocorticoid hormone) increases sodium reabsorption and potassium and H+ secretion in the cortical collecting tubule. Sodium retention expands ECF volume and decreases proximal tubule sodium reabsorption. Metabolic alkalosis then results from the combination of excess mineralocorticoid hormone effect and increased distal delivery of sodium (in the presence of aldosterone, sodium in the lumen will enter the cells of the collecting tubule and potassium and H+ will exit the cells into the tubule lumen). Administration of diuretic drugs that impair ion transport in the loop of Henle or distal convoluted tubule also results in stimulation of aldosterone secretion (secondary to renin release in response to hypovolemia) and increased sodium delivery to the collecting tubule.
Of note, high levels of aldosterone per se may not result in metabolic alkalosis in the absence of adequate distal sodium delivery. For instance, in congestive heart failure (CHF) and liver cirrhosis, there is secondary hyperaldosteronism (due to decreased renal perfusion and increased renin secretion); however, distal sodium delivery is decreased and metabolic alkalosis does not normally occur unless diuretics are administered.
• Shift into cells—severe K+ deficiency
With very severe potassium deficiency, K+ will shift out of cells into the ECF in exchange for H+. Therefore, H+ is “lost” into the cells, engendering metabolic alkalosis. The increase in intracellular H+ in renal tubular cells results in increased H+ secretion and bicarbonate reabsorption, thus also maintaining metabolic alkalosis (see below).
■ Net addition of HCO3− to ECF
■ Causes of HCO3− gain:
• Exogenous alkali administration (bicarbonate, lactate, citrate, acetate), especially in the presence of impaired renal function.
■ Loss of fluid containing chloride in excess of bicarbonate (“contraction alkalosis”)
■ Causes of Cl−-rich fluid loss:
• GI—villous adenoma, congenital chloridorrhea
 Villous adenomas are tumors that secrete chloride into the stool; K+ depletion also contributes to the generation of alkalosis. Congenital chloridorrhea is a rare disorder in which there is a failure of gut reabsorption of chloride secreted by the stomach.
Villous adenomas are tumors that secrete chloride into the stool; K+ depletion also contributes to the generation of alkalosis. Congenital chloridorrhea is a rare disorder in which there is a failure of gut reabsorption of chloride secreted by the stomach.
• Renal—diuretics, Bartter and Gitelman syndromes
 Diuretics are a very common cause of metabolic alkalosis because chloride reabsorption by the kidney is impaired and volume contraction results in stimulation of the renin–angiotensin–aldosterone axis (secondary hyperaldosteronism)
Diuretics are a very common cause of metabolic alkalosis because chloride reabsorption by the kidney is impaired and volume contraction results in stimulation of the renin–angiotensin–aldosterone axis (secondary hyperaldosteronism)
 Bartter syndrome is a genetic defect of the loop diuretic-sensitive Na–K–2Cl cotransporter in the loop of Henle (“endogenous loop diuretic”) (Simon, 1996). Gitelman syndrome is a genetic defect of the thiazide-sensitive Na–Cl cotransporter in the distal tubule (“endogenous thiazide”) (Monkawa et al., 2000) During chronic hypercapnia (seen frequently in chronic obstructive lung disease), there is an appropriate adaptive increase in renal H+ secretion and thus bicarbonate reabsorption. This is accompanied by loss of chloride in the urine (sodium is reabsorbed preferentially with bicarbonate rather than chloride). Rapid restoration of PCO2 to normal with mechanical ventilation is not accompanied by a similarly rapid change in bicarbonate handling by the kidney and may result in severe alkalemia. Because of previous chloride depletion, posthypercapnic metabolic alkalosis is typically associated with a low urine chloride concentration and improves with saline administration.
Bartter syndrome is a genetic defect of the loop diuretic-sensitive Na–K–2Cl cotransporter in the loop of Henle (“endogenous loop diuretic”) (Simon, 1996). Gitelman syndrome is a genetic defect of the thiazide-sensitive Na–Cl cotransporter in the distal tubule (“endogenous thiazide”) (Monkawa et al., 2000) During chronic hypercapnia (seen frequently in chronic obstructive lung disease), there is an appropriate adaptive increase in renal H+ secretion and thus bicarbonate reabsorption. This is accompanied by loss of chloride in the urine (sodium is reabsorbed preferentially with bicarbonate rather than chloride). Rapid restoration of PCO2 to normal with mechanical ventilation is not accompanied by a similarly rapid change in bicarbonate handling by the kidney and may result in severe alkalemia. Because of previous chloride depletion, posthypercapnic metabolic alkalosis is typically associated with a low urine chloride concentration and improves with saline administration.
• Skin—cystic fibrosis
Metabolic alkalosis has been described in children with cystic fibrosis due to loss of chloride in excess of bicarbonate in sweat.
Maintenance of Metabolic Alkalosis
Under normal physiologic conditions, bicarbonate is filtered by the glomerulus (the filtered load is the product of the glomerular filtration rate (GFR) and the plasma bicarbonate concentration). Virtually, all of the filtered bicarbonate is then reabsorbed, primarily at proximal nephron sites. Since normally about 1 mEq per kg of protons are generated by the body each day, this amount of bicarbonate buffer is titrated in the ECF and needs to be regenerated in the distal nephron. Both reabsorption (also termed “reclamation”) of filtered bicarbonate and regeneration of bicarbonate occur by tubular secretion of H+. H+ is formed in the tubular cell from the splitting of H2O into H+ and OH−. After the H+ is secreted into the tubular lumen, OH− then combines with CO2 to form HCO3−, which is reabsorbed into the peritubular capillary. Reabsorption of filtered bicarbonate occurs by titration of filtered bicarbonate by H+ in the tubular lumen and addition of bicarbonate formed in the tubular cell into the peritubular capillary. In the distal nephron, luminal bicarbonate is usually absent; therefore H+ secretion results in urinary H+ loss (primarily in the form of ammonium ions or NH4+) and thus bicarbonate regeneration (Fig 7.1).
Once metabolic alkalosis has occurred, its maintenance must indicate a failure of the kidneys to excrete the excess bicarbonate. This can occur either because of a decreased filtered load of bicarbonate (due to a decrease in GFR) or an increase in tubular bicarbonate reabsorption (or decrease in bicarbonate secretion). In the absence of renal failure, inability of the kidneys to excrete the excess bicarbonate implies the presence of a factor (or factors) that either increase bicarbonate reabsorption or decrease its secretion, such as ECF chloride depletion, potassium depletion, and hypercapnia. The mechanism(s) by which these factors maintain metabolic alkalosis are given below.
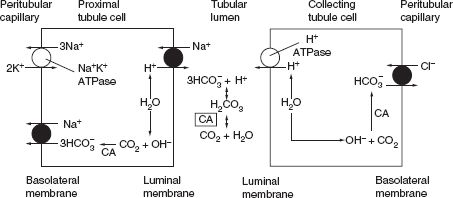
FIGURE 7.1 Mechanism of bicarbonate reabsorption and regeneration by tubular cells. The figure depicts the major cellular and luminal events in bicarbonate reabsorption in the proximal tubule (left panel) and regeneration in collecting tubules (right panel). In the proximal tubule, H+ ions are secreted into the lumen by the Na+–H+ exchanger, whereas HCO3− ions are returned to the systemic circulation primarily via an Na+–3HCO3− cotransporter. In the collecting tubule, an H+–ATPase pump and a Cl–HCO3− exchanger mediate these processes. In the proximal tubule, secreted H+ combines with filtered HCO3− to form CO2 and H2
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


