GISTs have evolved from poorly defined, histogenetically obscure GI mesenchymal tumors to well-defined oncogenic entities with distinctive clinical, morphologic, ultrastructural, histiogenetic, and molecular features, for which targeted therapy is available.
Demography and Clinical Aspects
Although rare, GISTs are the most common mesenchymal tumor of the GI tract. Population-based studies from the United States, Sweden, Iceland, the Netherlands, and Spain indicate that GIST has an annual incidence in the range of 6.5-14.5/million.
9,
10,
11,
12,
13 Up to 4,500 to 6,000 new cases are diagnosed per year in the United States.
4 GISTs occur mainly in adults with a median age of 55 to 60 years (over 90% occur after 40 years). The age-adjusted incidence for blacks is almost twice that of whites. GISTs show a slight male predominance in adults, while in the pediatric population females are more frequently affected.
12,
14 GISTs arise most commonly in the stomach (60%) and jejunum and ileum (30%) and less frequently in the duodenum (5%), colorectum (<5%), and esophagus (<1%). Rare sites include the omentum, mesentery, and retroperitoneum.
15 Case reports of GIST involving the gallbladder, liver, pancreas, and urinary bladder are on record.
16,
17,
18,
19GI bleeding (acute or insidious) is the most common presentation but GISTs may also present with bloating, early satiety, abdominal pain, palpable mass, obstructive symptoms, tumor rupture (rare), or incidentally at endoscopy or surgery. Most symptomatic patients present with GISTs that are larger than 5 cm in maximum dimension.
Approximately, 20% to 25% of gastric GISTs and 40% to 50% of small intestinal GISTs behave in a clinically malignant fashion. Metastases usually involve the abdominal cavity and liver. Rarely, the musculoskeletal system or skin may be involved, while metastasis to lymph nodes or lungs is extremely rare. Since metastasis can occur 10-15 years after initial presentation, long-term follow-up is required.
Complete surgical excision is the treatment of choice, but patients with unresectable or metastatic disease are treated with c-kit/PDGFRA tyrosine kinase inhibitors such as imatinib. Most patients show a partial or complete remission, but the development of resistance (due to secondary mutations or clonal selection) has limited the long-term efficacy of this therapy. In such cases, second-line tyrosine kinase inhibitors such as sunatinib may be used.
15,
20
Targeted Therapy with Imatinib
With the advent of molecularly targeted therapies and detailed identification of molecular signatures in GIST, a group of investigators from academia and industry developed and characterized signal transduction inhibitor (STI-571) or imatinib mesylate as a potent tyrosine kinase inhibitor of both c-kit and PDGF receptor (PDGFR).
24a Since then, there has been an extraordinary expansion in the clinical development of imatinib and similar class of inhibitors for molecularly targeted therapy of GIST.
Imatinib significantly improves clinical outcomes in patients with metastatic disease and in the postsurgical adjuvant setting. In addition, imatinib is often used in the preoperative setting to reduce tumor bulk to improve resectability and/or to avoid a more extensive surgical procedure with associated morbidity.
24b KIT and
PDGFRA mutational status is helpful in predicting response to imatinib and determining
the optimal dose of therapy (see Molecular Features). Primary resistance to imatinib is observed in about 15% GIST patients, but more than 80% will ultimately develop secondary resistance driven by additional KIT mutations.
24c Sunitinib maleate has been used as a second-line agent in imatinib-resistant patients. Sunitinib has also been used as first-line treatment in GISTs that are wild type for
KIT and
PDGFRA or that have mutations that are known to confer imatinib resistance. Although imatinib is quite well tolerated, long-term intake of sunitinib is complicated by significant adverse effects (fatigue, diarrhea, hand-foot syndrome, hypertension, and myelosuppression) in nearly 20% of patients.
24d Several other drugs are being tested for patients progressing on imatinib and sunitinib (e.g., regorafenib, sorafenib, dasatinib, nilotinib),
24c,
24d with regorafenib showing significant survival benefit in a randomized, placebo-controlled phase 3 trial.
24c
Gross Examination and Appearances
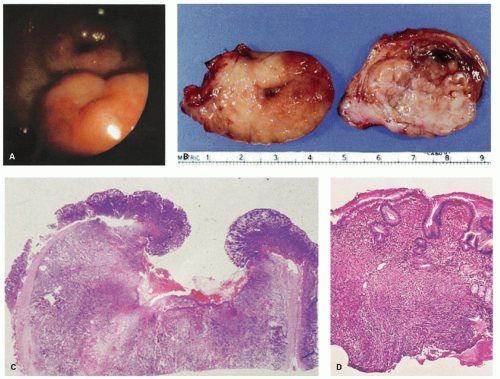
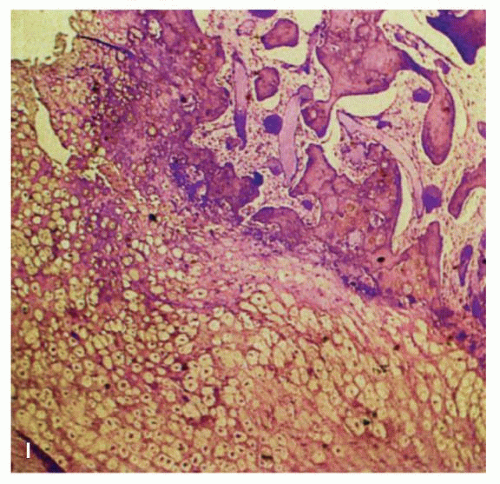
Gross examination should include careful inspection of the serosa for peritoneal breach. The mucosal surface should be examined for ulceration or tumor involvement. The maximum tumor dimension should be accurately recorded as this is a critical prognostic determinant. The serosa and resection margins should be inked and adequately sampled. Inspection of the cut surface will show most GISTs to be centered in the muscularis propria although they may extend inward toward the submucosa, outward toward the serosa, or both (
Fig. 7-2). A few have a characteristic dumbbell shape with tumor on either side of the muscularis propria. Some project into the lumen while others hang off the serosal surface. GISTs occasionally appear as omental or mesenteric nodules. GISTs vary in size from <1 cm to over 35 cm, with a median size of approximately 5 cm.
Small incidental GISTs have a predilection for the proximal stomach and gastroesophageal junction. Most GISTs are well circumscribed and nonencapsulated (
Fig. 7-2) but some compress surrounding muscle and connective tissue creating the impression of a capsule. GISTs generally lack the whorled appearance of leiomyomas and have a more homogenous, often lobu-lated, cut surface (
Fig. 7-2B). They vary from tan to red to brown. Cystic degeneration and hemorrhage are frequent (particularly in large tumors,
Fig. 7-2D-G), and necrosis may be present. Degenerative features do not indicate malignancy but are seen primarily in larger tumors, which are more likely to behave aggressively.
Most GISTs are solitary, but multifocal GISTs may be seen in association with tumor syndromes (see GIST syndromes) or sporadically.
2,
3,
4,
15,
23 At the time of gross examination of a suspected GIST, it is useful to freeze some fresh tissue in case
PDGFRA/
KIT mutational studies or molecular studies (to rule out certain GIST mimics—see later) are needed (see later); however, paraffin sections suffice in most cases.
Microscopic Appearances
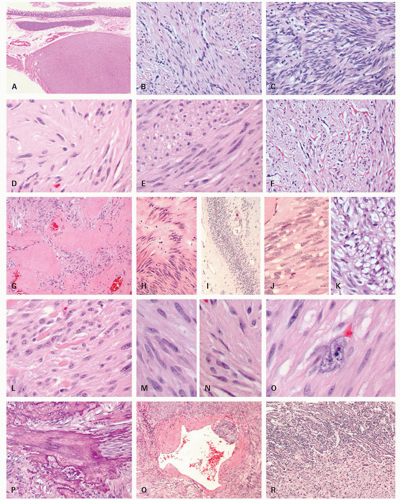
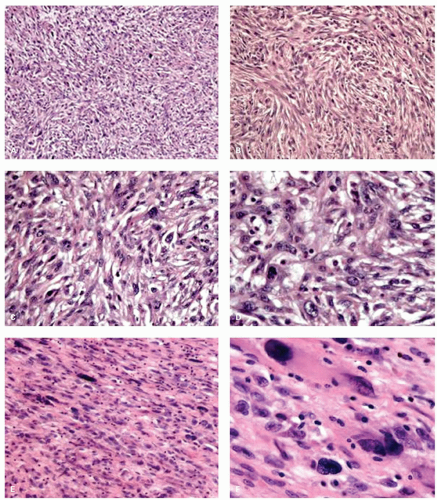
Histologically, GISTs may display spindle (70%), epithelioid (20%), mixed spindle and epithelioid (10%), and rarely pleomorphic morphology.
1,
2,
3,
4,
25,
26,
27 Detail of each morphologic subtype is provided in
Figures 7-3,
7-4 and
7-5.
Epithelioid morphology is seen in 40% of gastric GISTs but is rare in small intestinal GISTs (5%). Most GISTs are cytologically bland although focal cytologic atypia is not uncommon. Diffuse pleomorphism is distinctly unusual and should prompt consideration of alternative diagnoses (e.g., sarcoma, melanoma, or sarcomatoid carcinoma). Examples of pleomorphic GIST are shown in
Figure 7-5. The Armed Forces Institute of Pathology (AFIP) series of 1,765 gastric GISTs, the largest single site series to date, described eight distinctive histologic variants (four spindled and four epithelioid) with consistent and reproducible morphology
3 (
Fig. 7-6). Small intestinal GISTs could not be similarly subclassified.
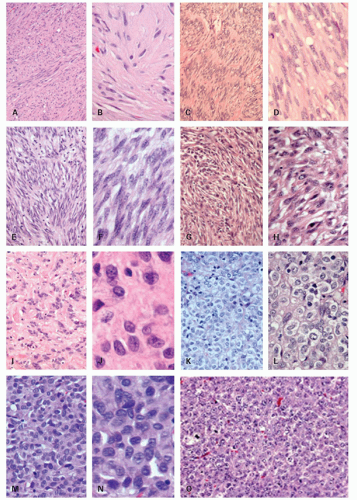
2Unusual morphologic variants. GISTs may rarely display unusual features that can lead to diagnostic confusion (
Fig. 7-7). GISTs may rarely show epithelioid differentiation to the point of gland formation,
28 prominent signet ring cell morphology (often merging with areas of more conventional GIST),
29 rhabdoid morphology including paranuclear whorls of vimentin filaments and often PDGFRA mutations
30 and rhabdomyomatous morphology.
17 Other variants include paraganglioma-like GISTs,
3 GISTs with prominent osetoclastic-like giant cells,
31 mesothelioma-like GIST (with epithelioid cord-like areas and pseudoglandular formations in a myxoid stroma), oncocytic variants (with abundant mitochondria), small cell variants (with crowded angulated nuclei), and cytotoxic T-lymphocyte-rich GIST (infiltrated by cytotoxic T cells).
Posttreatment changes. Histologic changes described postimatinib include those associated with tumor cell necrosis as well as phenotypic changes in residual tumor cells. Most GISTs show some histopathologic response to imatinib, but this is usually incomplete and does not correlate with clinical or radiologic response. Even tumors with a very good histologic response (i.e., >90% necrosis or hyalinization or both) show focal residual viable tumor in most cases if carefully looked for. Immunohistochemistry for CD117 or DOG1 may help to highlight such foci, although CD117 immunoreactivity, in particular, may be lost. DOG1 immunoreactivity tends to be better retained.
32
The histologic response often varies between and within individual nodules. Some tumors have a distinctive myxohyaline stroma, while others show extensive cystic change or hemorrhage. A small minority of tumors may show phenotypic alterations in residual tumor including switch from a spindled to epithelioid phenotype, small cell phenotype, acquisition of myoid phenotype (including desmin expression and ultrastructural features), and loss of CD117 and CD34 immunoreactivity.
Diagnostic problems may arise when secondary tumors with unusual morphology and unexpected immunohistochemical profiles arise at new locations. Mutational analysis may be extremely helpful in such circumstances.
33,
34,
35 The pathology report should include an estimate of the proportion of necrotic/hyalinized tumor as an indication of pathologic response. A grading system for pathologic response has been proposed, but this is not widely used outside the research setting.
36
Immunohistochemistry
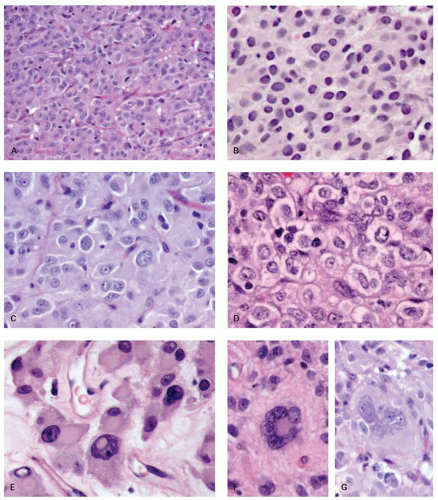
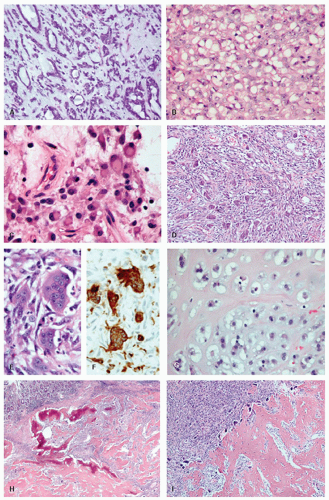
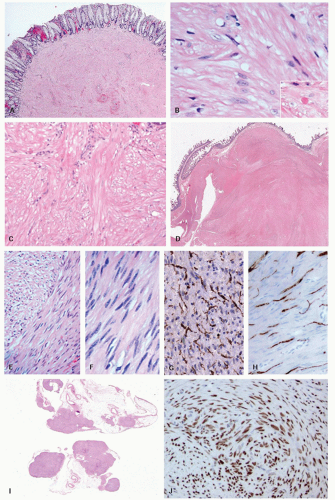
Approximately, 95% of GISTs are immunoreactive for CD117. Staining is usually strong and diffuse, most often cytoplasmic but can also be membranous or may involve the paranuclear “Golgi zone” with dot-like immunoreactivity (
Fig. 7-8A-D). Membrane staining is more often appreciated in epithelioid GISTs, while staining in spindle cell GISTs is typically pancytoplasmic; membrane staining may difficult to appreciate in spindled cells due to their narrower width. Epithelioid GISTs often show less uniform staining and are sometimes only weakly positive or negative (
Fig. 7-8E).
2,
3DOG1 (discovered on GIST 1) is a highly sensitive GIST marker, staining approximately 95% of these tumors. Originally identified in gene expression profiling studies, DOG1 is a calcium-activated chloride channel protein, also known as anoctamin 1 or TMEM16A. DOG1 shows a cytoplasmic and/or membranous pattern of staining similar to that seen with CD117.
37,
38 DOG1 appears to be a more specific marker of GIST than is CD117, although a handful of tumors other than GIST may occasionally express this protein (see below).
A number of alternative immunohistochemical markers for GIST have been evaluated, including nestin, protein kinase C theta, and PDGFRA. While nestin and protein kinase C theta have a high sensitivity, their expression by a wide range of histologic mimics of GIST limits their utility.
32 PDGFRA has been reported by some to be highly sensitive and generally specific for CD117-negative GISTs.
39,
40 However, the unpublished experience of most large GIST centers (including our own) is that reliability and reproducibility of commercial sources of this antibody are poor and therefore not suitable in the diagnostic setting.
41Other commonly expressed but nonspecific markers include CD34, nestin, Bcl2, and h-caldesmon. Smooth muscle actin (SMA) is expressed in a minority of cases (30%-40%), while S100 immunoreactivity is rare (5%-10%).
1,
4,
42,
43,
44,
45,
46 GISTs may occasionally show immunoreactivity for CK18 (4%-8%)
25,
47 and to a lesser extent CK8,
15 but are nonreactive for AE1/AE3.
48,
49 Expression of desmin is uncommon (<2%) and usually encountered in gastric epithelioid GISTs where expression is focal. Desmin may also be expressed following treatment with imatinib.
35 While interpretation of CD117 immunoreactivity is usually straightforward, significant challenges may arise particularly in small biopsy specimens. These challenges are outlined below together with practical measures to overcome them.
False-positive immunoreactivity. Overdiagnosis of GIST through misinterpretation of CD117 immunohistochemistry is more frequent than underdiagnosis due to failure to perform or correctly interpret CD117 immunohistochemistry. False-positive immunoreactivity is largely related to technical factors such as antibody dilutions, particular sources of commercial antibodies, and overvigorous antigen retrieval techniques, in particular heat-induced epitope retrieval (HIER).
1,
42,
50,
51,
52 A prime example is the early report of CD117 immunoreactivity (antibody source and dilution related) in desmoid tumors,
53 which under optimum technical conditions are CD117-negative tumors.
The use of HIER in CD117 immunohistochemistry is controversial and conflicting recommendations have been made regarding its use.
3,
10,
54 Irrespective of the method used, careful attention to positive controls (particularly ICC but also mast cells) and negative controls (especially smooth muscle and fibroblasts) is critical when establishing immunohistochemistry protocols (
Fig. 7-8F-I). For this purpose, we select blocks, wherever possible, that include muscularis propria and myenteric plexus and titrate out until any smooth muscle or fibroblast immunoreactivity has been quenched.
In practice, false-positive immunoreactivity is rarely a problem with resected tumors, where blocks containing in-built controls can be selected. The greatest difficulties arise with limited biopsy specimens, usually from metastatic sites. Here, the pattern of CD117 staining within cells, the distribution of staining in the biopsy, and profile of other markers (e.g., DOG1) need to be taken into account. Non-GISTs rarely show either membranous staining or paranuclear membrane accentuation. Rather, the staining is typically cytoplasmic and granular and usually focal and weak. If present, the staining of control material (usually mast cells) is much stronger. Nonspecific staining toward the edge of the biopsy specimen (fringe artifact) should be ignored and not interpreted as focal positive staining.
55 Other factors that weigh into the interpretation include tumor location, morphology, and the results of other immunostains (see above). If doubt regarding CD117 immunoreactivity persists, referral to a center with special expertise and/or mutational analyses and/or rebiopsy may help to resolve the diagnosis.
20,
55,
56CD117-negative GISTs. A small subset of tumors (approximately 5%) qualify as GISTs by all other criteria (including demographics, clinical presentation, anatomic location, morphology and immunohistochemical profile) but are negative for CD117 on immunohistochemistry. Lack of immunoreactivity may be due to technical factors (suboptimal fixation, excessive heat during section drying, or very prolonged storage of slides), sampling error (small biopsies from GISTs with only focal CD117 overexpression), loss of KIT overexpression following clonal evolution (e.g., after imatinib therapy), or genuine lack of CD117 overexpression.
56 Up to 50% of CD117-negative GISTs are immunoreactive for DOG1. A study of 1,040 GIST cases found only 2.6% of GISTs to be both CD117 and DOG1 negative. The true figure is likely to be even lower since two-thirds of CD117/DOG1- GIST that were tested for
KIT and
PDGFRA mutations were negative, challenging the original diagnosis. Two other studies have reported CD117-/DOG1- cases to comprise 0.9% and 1.6% of all GISTs.
57,
58
Up to 90% of CD117-negative tumors have mutations in either
PDGFRA (35%-80%) or
KIT (15%-20%).
4,
42,
59,
60,
61 Mutational analysis is therefore advocated where feasible in all CD117 tumors that otherwise have the typical features of GISTs.
42,
61 Turnaround time for this has improved dramatically beyond the point where it was easier to determine clinical response to imatinib than to wait for mutational analysis reports in overtly malignant tumors.
CD117- and DOG1-Positive Tumors Other Than GISTs
A number of tumors other than GISTs can express CD117 (
Table 7-2). Fortunately, most do not overlap morphologically with GISTs.
52 Those may include melanoma,
62 angiosarcoma,
63 endometrial stromal sarcoma,
64 PEComa,
65 and Kaposi’s sarcoma.
63 A small percentage (2%-4%) of colorectal adenocarcinomas may overexpress CD117
66,
67,
68. The figure of CD117 positivity is higher (10%-15%) for non-small cell carcinomas of the lung.
69 As melanoma can occur as both primary and polypoid metastases throughout the GI tract, this is the main issue with CD117 immunoreactive tumors. At least one melanoma marker (e.g., S100) should always be in a panel, and any immunoreactivity should lead to staining for additional markers, for example, HMB-45, MART-1, or tyrosinase.
Potential histologic mimics of GIST that may express DOG1 include Ewing’s sarcoma, glomus tumor, synovial sarcoma, angiosarcoma, leiomyosarcoma, peritoneal leiomyomatosis, uterine-type retroperitoneal leiomyoma, and desmoplastic melanoma. To date, there are only four GIST mimics that can express both CD117 and DOG1 (namely angiosarcoma, synovial sarcoma, Ewing’s sarcoma, and melanoma). In all four tumors, staining is focal with at least one of the markers. Thus, GIST remains the only mesenchymal neoplasm that can show diffuse immunoreactivity for both markers.
32 Therefore, in a morphologically typical GIST that shows diffuse immunoreactivity for CD117 and DOG1, no further immunohistochemical stains should be necessary.
Molecular Features and Mutational Analysis
Approximately, 80% of GISTs have mutations in
KIT, while about 5% to 7% have
PDGFRA mutations;
KIT and
PDGFRA mutations are mutually exclusive.
KIT mutations lead to constitutive upregulation of the protein tyrosine kinase, through ligand independent dimerization.
15,
70,
71 Most
KIT mutations occur in exon 11 (60%-70%), followed by exon 9 (10%-15%; almost exclusively small intestinal GIST), exon 13 (1%-4%), and exon 17 (<1%).
4,
42,
72 Very rarely, mutations have been identified in exons 8, 12, 14, and 18 in primary GIST.
73,
74 Exon 11 and 9 mutations cause dysregulation through alterations of the juxtamembrane autoinhibitory region of c-kit, while exon 13 and 17 mutations activate c-kit’s enzyme pocket directly.
PDGFRA mutations are strongly associated with gastric location and epithelioid phenotype.
2,
3,
59,
60,
75,
76 The most common is the D842V point mutation on occurring on exon 18, which comprises over 80%
PDGFRA mutations.
73The nature and site of
KIT and
PDGFRA mutations predict response to imatinib mesylate.
KIT mutations involving exon 11 are associated with a far better response to imatinib mesylate therapy than those involving exon 9.
42 The
PDGFRA exon 18 mutation (D842V) is associated with a poor response to imatinib, while GISTs with other
PDGFRA mutations are potentially imatinib sensitive.
61,
73,
75 GISTs lacking either
KIT or
PDGFRA are poorly responsive.
42 The occasional presence of
KIT mutations in melanoma
77 may represent a potential diagnostic pitfall.
KIT mutations may also be found in seminomas and systemic mastocytosis, they fortunately do not occur in tumors overlapping morphologically with GIST. Mutational studies performed on frozen tumor provide the best diagnostic yield. Although reasonable yields may also be obtained from DNA extracted from paraffin sections, this diminishes considerably with old archival material.
71 Areas of the slide containing tumor should be selected (using microdissection if necessary) to ensure a high proportion of tumor cells in the sample. Failure to do so may result in a false-negative result due to a high background of wild-type DNA.
Mutational analysis has considerable utility in a number of situations. It is particularly useful in the workup of suspected GISTs that are CD117 negative, since the vast majority have mutations in either
KIT or
PDGFRA genes.
4,
42,
61 It may also be helpful in predicting response to imatinib (see above) and determining the optimal dose of therapy. For example,
KIT exon 9 mutations are associated with a significantly longer disease-free survival when GISTs are treated with high-dose imatinib compared to standard dosages; such benefits are not observed in GISTs with other mutations.
78 Mutational status may also predict response to the second-line agent sunitinib as those harboring
KIT exon 9 mutations appear to be more sensitive to this agent than those harboring exon 11 mutations. The clinical benefit of sunitinib in wild-type cases has also been reported.
71 The type of mutation may also have prognostic implications. For example, gastric GISTs with
missense mutations in exon 11 seem to have a better prognosis than those with exon 11
deletions (no such association is found for small intestinal GISTs). Earlier reports of an unfavorable outcome for exon 9 mutations seem to be related to the poorer prognosis in small intestinal GISTs where these mutations are markedly overrepresented. At present, there are insufficient data for incorporation of mutational status into risk stratification schemes.
15,
71 Mutational analysis facilitates the diagnosis of familial GIST syndromes associated with germline mutations in
KIT or
PDGFRA. Here, both tumor and surrounding nontumorous tissue exhibit the same mutation. The demonstration of wild-type
KIT and
PDGFRA may provide indirect support for the diagnosis of neurofibromatosis 1 (NF1) or Carney’s-associated GISTs in the right clinical setting (see below). Mutational studies can demonstrate secondary
KIT mutations associated with the onset of secondary resistance to imatinib. Finally, mutational analysis can distinguish multiple sporadic GISTs (showing distinct
KIT mutations in different tumors) from metastases or multiple familial GIST, which share a common mutation (see Sporadic multiple GIST below).
There are differing views as to whether mutational analysis should be a part of the routine diagnostic workup of all GISTs.
41 Most would argue that this is not justified at present and should be reserved for specific situations such as those outlined above, or when contemplating neoadjuvant therapy in unresectable tumors. However, mutational studies should continue to be performed in the research setting particularly in studies evaluating new therapies.
Succinyl dehydrogenase subunit B (SDHB) expression, NF1 mutations, and BRAF mutations. Of the 10% to 15% of GISTs that are wild type for
KIT or
PDGFRA, a proportion will show other molecular abnormalities, including loss of succinyl dehydrogenase subunit B (SDHB) expression, NF1 mutations, and occasionally BRAF mutations (the latter reported in 7%-11% of wild-type GIST).
32,
79,
80,
81,
82 Mutations in any one of the SDH subunits A to D can result in loss of immunohistochemical expression of SDHB.
83
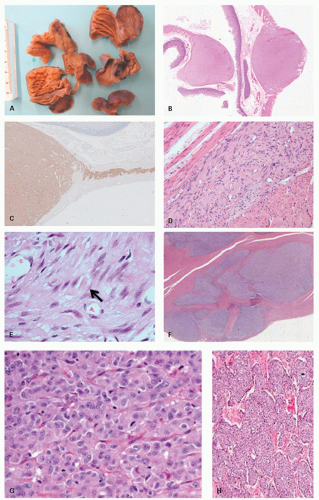
SDHB-deficient GISTs. SDHB-deficient GISTs have distinctive clinicopathologic features. They are seen almost exclusively in children or young adults, have a female predominance, have a gastric location, are frequently multicentric and/or multinodular (often with a plexiform growth pattern), have epithelioid morphology, and frequently display lymphovascular invasion (
Fig. 7-9F,G). Despite a high frequency of liver and lymph node metastasis, survival is disproportionately long compared to unselected metastatic GISTs, and clinical behavior is
not predicted by size and mitotic rate.
81,
83,
84 SDHB-deficient GISTs are resistant to imatinib but may show a greater sensitivity to the second-line agent sunitinib than do
KIT– and
PDGFRA-mutated GISTs.
81
Four clinical subgroups are strongly associated with SDHB-deficient GISTs, namely Carney’s triad, Carney-Stratakis syndrome, pediatric GIST (see below), and a proportion of sporadic GIST in young adults. In an unselected series of 746 gastric GISTs, 7.5% were found to be SDHB deficient.
81 SDHB-deficient GISTs have been shown to overexpress insulin-like growth factor receptor 1 (IGFR1).
84 Thus, it could be argued that GISTs with classical clinicopathologic features and an SDHB−/IGFR1+ immunoprofile do not require the more costly
KIT and
PDGFRA mutational analysis.
Chromosomal alterations. While
KIT and
PDGFA mutations are the earliest genetic changes in most GISTs, chromosomal losses (and gains) accumulate with progression to more aggressive phenotypes. Most low/very low malignant potential GISTs have a normal karyotype or loss of 14q (the latter occurs in 2/3 of GISTs overall). Intermediate malignant potential GISTs may show further loss of 22q (present in 1/2 of GISTs overall) and/or loss of lp. Overtly malignant GISTs usually show further chromosomal loss (such as 9p, 11p, 13, 15, or 18) or gains (such as 5p, 8q, 17q, or 20q). Of interest, chromosome 9p21 contains the tumor suppressor gene
CDKN2A, which encodes the cell cycle inhibitors p16
INK4A and p14
ARF. Immunohistochemical and comparative genomic hybridization studies have shown p16 loss in GIST to be associated with a poor prognosis.
70,
71,
85
Predicting Behavior
The presence of histologically confirmed metastases clearly indicates a malignant tumor. It is good practice to histologically confirm the nature of suspected metastases found at surgery by biopsy; rarely these prove to be from a different tumor that demands different therapy. Such “metastases” may occasionally prove to be unrelated benign lesions (e.g., focal nodular hyperplasia in the liver, granulomata, or Von-Meyenburg complexes). Biopsy eliminates such uncertainties. Stromal tumors are also lethal by virtue of their local recurrence and invasion of other organs. Incomplete resection clearly indicates an increased likelihood of local recurrence even in the absence of metastases.
23 Tumor rupture at presentation or at surgery is independently associated with a poor outcome; fortunately, this complication occurs in <5% of patients.
86,
87A variety of prognostic factors have been evaluated in localized, surgically resectable GIST. These including tumor size, mitotic rate, site, tumor rupture, serosal involvement, mucosal ulceration, histologic type or pattern, lamina propria infiltration, coagulative necrosis, cellularity, degree of atypia, Ki67/MIB-1 index, p53, p16, ploidy, and others. Of these, tumor size and mitotic rate have consistently emerged as important independent prognostic factors. The significance of other factors has been difficult to ascertain due to small cohort size, selection bias, and association with other high-risk factors.
87 The NIH consensus classification system—based on a GIST workshop in 2001—stratified risk of aggressive behavior on the basis of size and mitotic rate (
Table 7-3).
88 This system was based on consensus opinion rather than distinct data sets but accumulating evidence has confirmed its prognostic utility. Two very large studies from the
AFIP including 1,765 gastric GISTs (1,074 with full long-term follow-up data; 48% alive and disease free at a median of 14.1 years) and 906 small intestinal GISTs (629 with full long-term follow-up data; 34% alive and disease free at median of 15.5 years) have shown tumor site to be an important independent predictor of prognosis,
2,
3 confirming the findings of previous smaller studies.
89,
90,
91Failure to account for site may result in significant overestimation of the risk of aggressive behavior in gastric GIST using the NIH 2001 risk stratification scheme. For example, while NIH scheme would place gastric GISTs >10 cm in size and with <5 mitoses/HPF into the high risk category, the AFIP study found that only 11% of these patients died of their GISTs during long-term follow-up. Other adverse prognostic features in the AFIP series included coagulative necrosis, mucosal invasion, and ulceration (gastric and small intestinal GISTs); proximal location in stomach versus antrum and exon 11 deletions versus point mutations (gastric GISTs); and diffuse cytologic atypia and epithelioid cytology (small intestinal GISTs). The authors have proposed a new set of guidelines with separate criteria for gastric and small intestinal GISTs (modified in
Table 7-4). The currently available data on GISTs at uncommon sites such as esophagus, colon, and rectum, although limited, suggest that these should be stratified similar to small intestinal GISTs.
15 Proliferation markers might be useful in assessing the proliferative rate but have not proved superior to mitotic rate in predicting aggressive behavior. As noted earlier, the behavior of SDHB-deficient GIST (which are exclusively gastric) is
not predicted by size and mitotic rate.
Mitotic rate counting may be subject to number of influences including interobserver variability, specimen fixation, section thickness, size of fields examined, variation in tumor cellularity and tumor cell size, and fastidiousness of the pathologist, all of which may affect reproducibility. If modern wide-field eyepieces are used, the mitotic rate should be adjusted to correspond to the same total area examined in the largest (AFIP) studies on which prognostic data are based (i.e., 5 mm
2). Counts should be performed in the most mitotically active area and should proceed until either 50 fields are viewed or 100 mitoses have been counted.
2,
3 Tissue should be adequately fixed and sections should not exceed 4 to 5
µm in thickness. Care should be taken not to count pyknotic nuclei or nuclear debris as mitotic figures. Two studies have stratified GISTs into high- and low-risk groups based on gene expression profiles and chromosome complexity, both outperforming the AFIP grading system.
92,
93 It is therefore possible that morphology-based risk stratification schemes might ultimately be replaced by risk stratification based on a molecular profiling.
GIST Syndromes
Four GIST syndromes have been described: (1) familial GIST syndrome, (2) neurofibromatosis type 1 (von Recklinghausen’s disease), (3) Carney’s triad, and (4) GIST paraganglioma syndrome (
Table 7-5).
Familial GIST syndrome. Familial GIST syndrome, associated with germline mutations in
KIT94,
95,
96,
97,
98,
99,
100 or
PDGFRA101 genes, is extremely rare with at least 25 kindreds reported.
102,
103 Those affected have several family members who develop multiple primary
GISTs at early age (on average two decades earlier than sporadic GISTs) (
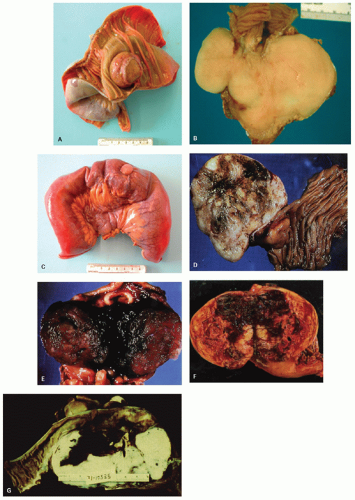
Fig. 7-9A-E). GISTs typically develop in a background of diffuse ICC hyperplasia. Depending on the mutation involved, patients may develop a variety of cutaneous lesions (including hyperpigmentation of per oral, axillary, perineal regions and hands, lentigines, café au lait macules, benign nevi, urticaria pigmentosa, and melanoma), hematologic malignancies, or dysphagia (presumably due to dysmotility associated with ICC hyperplasia) (
Table 7-5). There is an autosomal dominant pattern of inheritance.
42,
94,
95,
96,
97,
98,
99,
100,
101 In this setting, multiple primary GISTs are distinguished from metastases on the basis of their small size, lack of mitotic activity, and uneventful follow-up (unless associated with a concurrent malignant tumor).
2
Neurofibromatosis type 1 (von Recklinghausen’s disease). Patients with NF1 are at increased risk of the development of GISTs. A Swedish study found the incidence of clinically apparent GISTs to be approximately 7% in NF1 patients and 33% in an autopsy series.
104 NF1 is overrepresented by 50- to 150-fold among patients with GISTs.
10,
105,
106 GISTs associated with NF1 are frequently multicentric (60%) and often accompanied by ICC hyperplasia. The vast majority occurs in the small intestine (usually jejunum) with the stomach infrequently involved.
105,
106,
107,
108 On average, NF1-associated GISTs present approximately 10 years earlier than sporadic GISTs.
106 Most GISTs occurring in the setting of NF1 have low-grade morphology, low mitotic rates, abundant skenoid fibers, and a good clinical outcome,
4,
105,
106,
109 but a minority may show more aggressive behavior.
106,
108 Despite CD117 overexpression, almost all lack
KIT or
PDGFRA mutations.
105,
106,
107 NF1-associated GISTs show diffuse S100 immunoreactivity in up to 40% to 60% of cases.
106,
109 Occasionally, GISTs may be the first presentation in clinically unrecognized NF1.
110 The pathologist should be alert to this possibility when confronted with multiple GIST, ICC hyperplasia, strong S100 immunoreactivity, and absence of
KIT or
PDGFRA mutations.
Carney’s triad. Carney’s triad is a tumor syndrome characterized by multicentric, functioning extraintestinal paraganglioma, pulmonary chondroma, and
gastric epitheliod GIST (
Fig. 7-9F-I). Gastric GIST may precede the other tumors by several years. About 80 cases have been reported in the literature to date.
111,
112,
113 Most are sporadic. Distinctive clinicopathologic features include a striking female predominance (>85% of cases), young age at presentation (>80% of patients <30 years), frequent multifocality, consistent epithelioid morphology, and a relatively indolent course despite a high rate of local recurrence and metastases (41% and 55% respectively).
111,
112 These GISTs also lack
KIT/
PDGFRA mutations as well as the nonrandom loss of 14q and 22q, characteristic of sporadic GIST. The response to imatinib in this setting is generally considered to be poor.
114
GIST-paraganglioma syndrome (Carney-Stratakis syndrome). This novel familial GIST syndrome (involving 12 patients from 7 families) overlaps with Carney’s triad in that patients develop both paragangliomas and GISTs at a young age.
115,
116 However, it differs from Carney’s triad in that there is an equal sex predilection, an absence of pulmonary chondromas, and an autosomal dominant pattern of inheritance with incomplete penetrance.
115 The majority of patients with this syndrome have germline loss-of-function mutations in the succinyl dehydrogenase subunit genes
SDHB, SDHC, and
SDHD,
116 leading to loss of SDHB expression and overexpression of IGF1R.
Sporadic multiple GIST. Multiple primary GISTs were thought to occur only in the clinical syndromes described above. However, a total of 21 patients with multiple primary sporadic GISTs (with differing
KIT or
PDGFRA mutations in each lesion) have been described in four studies.
117,
118,
119,
120 In 19 patients, the tumors arose in the same organ (16 stomach, 3 small intestine), while in 2 cases they involved different organs (stomach and small intestine, small intestine and peritoneum). Most were situated within 3 cm of one another. The mean age at presentation was 72.4 years. In about half of the cases, at least one of the lesions was under 1 cm in size. However, in one study five patients had two separate lesions >1.5 cm size (mean of 4.3 cm).
118 Sporadic multiple GISTs differ from those associated with GIST syndromes in that patients are older, have fewer GISTs (usually 2-3), lack ICC hyperplasia, often have smaller tumors, show a predilection for the proximal stomach (versus small bowel for familial and NF1-associated GISTs), are usually closely approximated (<3 cm), and show different
KIT or
PDGFRA mutations in each tumor. The existence of multiple sporadic GISTs has important clinical implications. Patients presenting with multiple GISTs in the absence of clinicopathologic features of GIST syndromes may be misdiagnosed as having metastatic disease (as occurred in 2/6 patients in one study) and treated with imatinib.
118 Molecular testing of each tumor is therefore recommended in this setting.
Pediatric GIST. Very occasionally, GISTs may present in children or young adults. Pediatric GISTs display a number of characteristic clinicopathologic features including a striking female predominance, a predominantly gastric location, a strong tendency for multifocality, a frequent epithelioid morphology, and high rate of local recurrence and lymph node metastasis.
81,
121,
122,
123 Almost all reported pediatric GISTs so far are negative for
KIT or
PDGFRA mutations,
122,
123,
124 although an isolated case with a novel exon 9
KIT mutation was reported.
124
Small GIST. Small, clinically silent GISTs are being detected with increasing frequency,
125 raising questions as to their clinical significance and management. Several studies suggest a high prevalence in the general population. Agaimy et al. detected small GIST in 22.5% of 98 consecutive autopsy cases by gross examination (this included only grossly recognizable lesions).
126 Kawanowa et al. detected microscopic GIST in 35 out of 100 gastrectomies (performed for unrelated tumors) when sectioned at 5 mm intervals.
127 Two studies of esophagogastrectomy specimens found microscopic GIST or minute lesions (≤1 mm) termed “ICC hyperplasia” in approximately 10% of patients
128,
129; good criteria for separating focal ICC hyperplasia and microscopic GIST do not exist. Small GISTs show a predilection for the proximal stomach and gastroesophageal junction. Most are solitary but up to 30% may be multiple. Reported cases have ranged from GISTs range from 0.2 mm in size to up
to 10 mm. The lesions frequently show hyalinization and calcification leading to the suggestion that many may regress/involute.
126 The reported rates of
KIT and
PDGFRA mutations vary but they occur in up to 85% of GISTs ≤10 mm in size.
130 Variable terminology has been applied to these lesions (microscopic GIST, minimal GIST, minute GISTs, GIST tumorlets, ICC hyperplasia, etc.), and there is no consensus on terminology. The simplest approach might be to regard all GIST-like lesions ≤10 mm in size as “small, incidental GIST” given their uniformly indolent behavior. The vast majority either involute or remain asymptomatic. A small minority may accrue additional genetic alterations and evolve into clinically significant GIST.

 Disorders of Endocrine Cells
Disorders of Endocrine Cells
 Gastrointestinal Manifestations of Extraintestinal Disorders and Systemic Disease
Gastrointestinal Manifestations of Extraintestinal Disorders and Systemic Disease
 Inflammatory Disorders of the Esophagus: Reflux and Nonreflux Types
Inflammatory Disorders of the Esophagus: Reflux and Nonreflux Types
 Small Bowel Mucosal Disease
Small Bowel Mucosal Disease
 Stomach and Proximal Duodenum: Inflammatory and Miscellaneous Disorders
Stomach and Proximal Duodenum: Inflammatory and Miscellaneous Disorders
 Small and Large Bowel Polyps and Tumors
Small and Large Bowel Polyps and Tumors



