Fig. 11.1
Membranous nephropathy. (a–b) Light microscopy showing thickened glomerular basement with pin holes and spikes ((a) periodic acid-Schiff stain, (b) silver methenamine stain, both 40×). (c) Immunofluorescence microscopy showing granular IgG along the capillary walls (20×). (d) Electron microscopy showing numerous subepithelial deposits (black arrows). Note basement membrane material between the deposits (white arrows) forming the spikes (6800×)
Although it may be difficult to determine whether the MN is primary or secondary, there are some features that are suggestive of secondary MN. Secondary MN should be suspected when the kidney biopsy shows (a) proliferative features – mesangial or endocapillary, (b) full-house pattern of Ig staining including staining for C1q on immunofluorescence microscopy, (c) electron dense deposits in the subendothelial location of the capillary wall and/or mesangium, or along the tubular basement membrane and vessel walls, and (d) endothelial tubuloreticular inclusions on EM [1]. A drug-associated or malignancy-associated secondary MN may be suggested by the presence of only a few superficial scattered subepithelial deposits on EM. A study by Yoshimoto et al. suggested that the location of the subepithelial deposits, i.e., subepithelial (homogeneous), with subgroups superficial and deep, versus subepithelial and intramembranous (heterogeneous) deposits is important in predicting prognosis: the homogeneous group with superficial deposits appears to predict a better prognosis versus the deep deposits and the heterogeneous group [2].
Finally, staining for IgG subclasses may also help to differentiate between primary from secondary MN. IgG1, IgG2, and IgG3 tend to be highly expressed in lupus membranous nephropathy (class V lupus nephritis), while IgG1 and IgG4 tend to be highly expressed in primary MN [3]. In addition, IgG4 tends to be absent in the immune deposits of MN secondary to malignancy [4].
11.3 Identification of Target Antigens in Membranous Nephropathy
Our understanding of the disease process in MN has evolved significantly over the past five decades. Glomerular disorders associated with immune deposits, such as MN, lupus nephritis, or post-infectious glomerulonephritis, were hypothesized to be universally due to the deposition of circulating complexes of immunoglobulin and antigen within the glomerulus. It was not initially clear why the deposits in idiopathic MN should be associated exclusively with the abluminal (subepithelial) side of the GBM, but mechanisms were postulated that invoked the dissociation and reassociation of circulating immune complexes (CIC) with appropriate physicochemical properties to traverse the negatively charged GBM [5].
Much of our initial understanding of the pathogenesis of MN has come from an experimental rat model of disease introduced by Walter Heymann in the late 1950s [6]. This model is eponymously known as Heymann nephritis and continues to be used to this day as the foremost existing animal model of MN. Animals are actively immunized with an antigenic fraction of rat tubular brush border membranes known as Fx1A, and several weeks later develop histologic and clinical features virtually identical to the human disease. A passive model, in which anti-Fx1A antibodies were first raised in sheep and subsequently injected into rats, also produced disease within days instead of the weeks required in the active model. Two independent research groups used this model to demonstrate that anti-Fx1A antibodies could induce disease in an isolated perfused rat kidney in which the antibodies had no chance to recirculate and/or form CIC [7, 8]. The formation of these early subepithelial immune deposits in the absence of CIC strongly suggested that the deposits formed in situ and that there might be an endogenous antigen present within the GBM or even on the podocyte cell membrane.
Subsequent work over the next decade identified the major target antigen within Fx1A as the large endocytic receptor megalin and demonstrated its presence not only in tubular brush border – the source material for Fx1A – but also on the podocyte [9, 10]. Ultrastructural immunoperoxidase studies revealed binding of the infused antibodies to megalin that was present at the surface and within clathrin-coated pits at the basal surface of the rat podocyte [11]. These antigen-antibody complexes were eventually enriched and shed as immune complexes immediately beneath the podocyte. Concurrent work, which will be detailed later in this chapter, implicated the complement system in the damage that occurs to the podocyte.
The identification of megalin as the target antigen in a rat model of MN begged the question of whether megalin or a related molecule might also be the target in human disease. Humans, as opposed to rats, were found to lack megalin expression in the podocyte, and thus megalin and a related molecule, the LDL related receptor, were quickly ruled out as antigenic targets in human disease. In this manner, the hunt for the true human MN target antigen(s) began.
It was not until 2002 that a relevant human target antigen was convincingly identified [12]. This occurred not in the common idiopathic form of MN that occurs in adults but rather in a rare alloimmune form of MN that has only been described in newborns. Mothers who were genetically deficient in the enzyme neutral endopeptidase (NEP, also called neprilysin) and who had been alloimmunized against this protein from a previous pregnancy that had been miscarried could give birth in a subsequent pregnancy to an infant with the nephrotic syndrome [12]. Biopsy of such an infant revealed MN, and it was shown that both the mother and the infant (due to transplacental transfer of immunoglobulins) had circulating antibodies to NEP. Because these maternal antibodies were eventually cleared, the MN and nephrotic syndrome was short lived in the newborn. This landmark study established NEP as the first identified antigen in human MN.
Several groups interested in this topic, including our own, continued to search for relevant human target antigens in adult MN. The experimental approach was straightforward to use the serum of patients with active MN to screen for reactivity against human glomerular antigens: by Western blotting. Despite such apparent simplicity, it was difficult to find bands that were consistently identified by any more than a few patient samples. In our experience, several years were spent partially purifying a protein band identified by at most three patients with MN and was ultimately not identified using these or other techniques.
A change in methodology provided the breakthrough in identifying the target antigen in adult MN. Western blotting is traditionally performed under conditions that allow proteins to assume their primary structure: they are boiled in the presence of detergent as well as agents that disrupt secondary structure due to disulfide bonding. Our laboratory made the decision to run the blots in the absence of reducing agent, leaving disulfide bonds intact, and immediately started to note the appearance of a high molecular weight band in the 160–180 kDa range but only when blotted with serum from MN patients. We also visualized these reactive autoantibodies using detecting antibodies specific for the IgG4 subclass of human IgG, which is the least abundant subclass in general but the predominant subclass of autoantibodies in idiopathic MN. This allowed for a higher signal-to-noise ratio than when we detected total IgG and thus allowed a very sensitive detection of what we then called the “MN antigen.”
Partial purification of this antigenic band proved somewhat difficult, but early recognition that it was a glycoprotein allowed us to capture it using wheat germ lectin column chromatography and then to strip off N-linked sugar molecules using the enzyme peptide N-glycosidase. When both fully glycosylated and deglycosylated forms were electrophoresed and the individual bands cut out and sent for mass spectrometric analysis, a limited number of transmembrane glycoproteins were identified. Well-known podocyte proteins such as nephrin or the alpha3 integrin chain were initially selected as candidate antigens but turned out negative. A second approach, in which the antigen was concentrated in membrane fragments or vesicles shed from living glomeruli into saline, followed by the same mass spectrometric approach, revealed a new protein that yielded the most abundant peptide spectra, the M-type phospholipase A2 receptor (PLA2R). A quick literature search identified Gérard Lambeau at the University of Nice as the investigator who had initially identified this protein in rabbit muscle and had later cloned the human gene [13]. Probing the recombinant protein with human sera quickly demonstrated that PLA2R was indeed the target antigen, and anti-PLA2R antibodies raised in guinea pigs by the Lambeau laboratory confirmed that PLA2R was a protein restricted in the kidney to the podocyte. The first antigen in adult MN was thus identified [14].
11.4 The M-type Phospholipase A2 Receptor (PLA2R)
The M (muscle)-type phospholipase A2 receptor is a member of the mannose receptor family and is composed of an N-terminal cysteine-rich (ricin B) domain, a fibronectin-like II domain, eight C-type lectin-like domains (CTLD), a transmembrane region, and a short cytoplasmic tail containing motifs used in endocytic recycling (Fig. 11.2). It was cloned based on its ability to bind secreted phospholipase A2 molecules, but its exact cellular function is not fully known. Previously unsuspected biological roles for PLA2R have recently been identified in nonrenal cells, which include promotion of replicative senescence in human dermal fibroblasts [15] and as a tumor suppressor in mammary epithelium [16]. It is tempting to speculate that PLA2R may also play a role in the post-mitotic, differentiated state of the podocyte, but its function in the kidney remains undefined at this point.
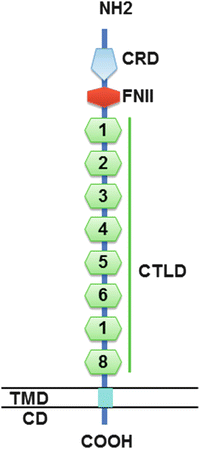
Fig. 11.2
Structure of PLA2R. The amino acid sequence of PLA2R starts with a 20-amino-acid signal peptide, followed by a large extracellular segment, a transmembrane domain, and a short intracellular domain containing an endocytosis motif. The extracellular segment contains a cystine-rich domain at the N-terminal, followed toward the membrane surface by a fibronectin type II domain and eight C-type lectin-like domains/carbohydrate recognition. CD, intracellular domain; COOH, intracellular C-terminal end; CRD, cysteine-rich domain (thin vertical lines, S-S bounds); FNII, fibronectin type II domain; CTLD, C-type lectin-like domain; NH2, extracellular N-terminal end; TMD, transmembrane domain (Modified from Ref. [32] with permission)
Recent work has identified the regions of the molecule that harbor the humoral epitopes targeted by anti-PLA2R autoantibodies. Using a series of PLA2R truncation mutants, Kao and colleagues demonstrated that the N-terminal region spanning from the cysteine-rich/ricin B (CysR) domain through the first CTLD contained the dominant epitope [17]. Fresquet et al. were able to narrow down the epitope to a 31-amino-acid stretch within CysR, which importantly contains a disulfide bond that appears to stabilize the epitope [18]. Work in our laboratory as well as the Lambeau laboratory has demonstrated that there exist at least three distinct humoral epitopes within PLA2R: a dominant epitope in CysR, with additional epitopes in CTLD1 and in the C-terminal half of the extracellular domain (unpublished observations). Identification of these epitopes may help in our understanding of the origins of this autoimmune disease (e.g., via molecular mimicry) or aid in therapeutic approaches that may block or otherwise immunoabsorb pathogenic autoantibodies.
11.5 Pathogenesis of Membranous Nephropathy
As described earlier, what we know about the pathogenesis of MN has largely been derived from studies in the Heymann nephritis experimental model of MN. The activation of complement plays a central role in the podocyte injury and the resulting proteinuria, since passive administration of only the gamma2 (non-complement fixing) fraction of anti-Fx1A or depletion of complement components with cobra venom factor did not cause podocyte injury or proteinuria, despite the formation of subepithelial immune complexes [19]. It appears that the aggregation of complement-fixing antibodies and antigen (megalin in the case of Heymann nephritis and likely PLA2R in humans) within the immune deposits is necessary for the local activation of complement. Despite the proteolytic generation of C3a and C5a during activation of the complement cascade, these anaphylatoxins are likely lost into the urine due to fluid flux into Bowman’s space and therefore are not able to recruit inflammatory cells from the circulation. This explains why MN is a noninflammatory glomerular disease. However, C5b with its other fluid phase partners can assemble into the C5b-9 membrane attack complex, which inserts into and thereby permeabilizes the podocyte cell membrane, allowing an influx of calcium that sets off a number of maladaptive intracellular signaling events that ultimately lead to simplification of the podocyte cytoskeleton, foot process effacement, and proteinuria (for review, see [20]). The podocyte is only sublethally injured and can shed membrane components containing antigen, antibody, and complement components. However, this injurious process continues as long as circulating antibody is available to target the relevant podocyte antigen.
It is quite likely that a similar pathogenic process occurs in human disease, but there are also several reasons to expect differences. There is clearly evidence of complement activation in human MN. One of the histopathologic hallmarks of MN is a fine granular capillary wall pattern of C3 deposits, in the same distribution as IgG. Although not typically assessed in MN, C4 is also present in the same pattern [21]. C5b-9 has been found both within close proximity of the immune deposits and in the urine (likely representing shed membrane from the injured podocytes) [22]. What is typically absent or present at low levels in primary MN is complement component C1q, a marker of the classical pathway of complement activation. Thus, within the subepithelial immune deposits of MN, there are antigen, IgG, complement factors C3 and C4 but limited evidence of the particular pathway by which the complement cascade is activated.
The predominant IgG subclass found within the subepithelial deposits of primary (but not secondary forms of) MN is IgG4, as noted above. IgG4 represents a rather unusual form of IgG in that it does not activate the classical complement pathway and is unable to form large immune complexes due to physiological separation and exchange of the half molecules (Fd fragments). Several hypotheses have been proposed to reconcile the presence of IgG4 and the products of complement activation. The first is that early in the disease course, there may be relatively more of the complement-fixing subclasses of autoantibody, namely, IgG1 and IgG3. Huang and colleagues have demonstrated in MN biopsy specimens that in early (stage I) disease, there tends to be more IgG1 within deposits and that IgG4 becomes predominant in later stages [23].
On the other hand, our recent study on recurrent MN post-kidney transplant (KTX) diagnosed by protocol biopsy challenges such observations that would otherwise implicate IgG1 or other non-IgG4 subclasses in early disease [24]. We have performed IgG subtyping on post-KTX biopsies in 13 patients who experienced recurrence of MN in the allograft. Of the seven patients (regardless of PLA2R status) that had biopsies available for IgG subtyping at the first time point at which histologic recurrence was noted, six out of these seven patients had exclusive or predominant IgG4 staining in capillary walls. Similarly, IgG4 was the only or dominant subclass in most of the earliest biopsies of subjects with PLA2R-associated MN. A single case of known non-PLA2R-associated MN was IgG1 predominant. Though our findings may be considered inconsistent with Huang et al., it should be pointed out that they evaluated IgG subclass staining in native kidney biopsies, whereas our study investigated recurrent MN in the allograft, which could be affected by transplant immunosuppression or previously class-switched memory B cells.
Preliminary evidence from the Beck and Salant laboratories has suggested that IgG4 may directly activate the lectin pathway of complement through the binding of mannan-binding lectin (MBL) to IgG4 molecules with hypogalactosylated glycan moieties [25]. Patients with MN, especially older patients, may have hypogalactosylated glycan chains attached to the IgG4 molecules that can bind MBL and activate this third pathway of complement activation. Furthermore, our recent study evaluating the role of C4d as a diagnostic tool in proliferative glomerulonephritis shows that immunofluorescence examination of native kidney biopsy from patients with MN is characterized by the absence of C1q but positive staining for C3 and C4d [26]. Taking this all together, the most attractive hypothesis is that in MN IgG4 may directly stimulate the lectin pathway of complement activation [27].
However, it should also be noted that IgG4 is not necessary to produce all the clinical and histopathologic features of MN. Debiec and colleagues have described a very rare case of recurrent MN in which both the recurrent and native diseases were caused by the presence of a monoclonal IgG3 kappa anti-PLA2R [28]. The only difference between this case and the usual findings in primary MN was the presence of strong C1q staining, which is understandable since IgG3 can activate the classical complement pathway. As such, there may be several pathways in MN by which complement can be activated to cause podocyte injury.
11.6 Genetic Associations
Initial studies from Korea and Taiwan showed that single nucleotide polymorphisms (SNP) in the phospholipase A2 receptor gene were associated with genetic susceptibility to MN [29, 30]. Subsequently, Stanescu et al., using a genome-wide association study of SNPs in patients with idiopathic MN from three different Caucasian population (75 French, 146 Dutch, and 335 British patients), identified significant alleles at two genomic loci associated with idiopathic MN: chromosome 2q24 containing the gene encoding PLA2R and chromosome 6p21 containing the gene encoding HLA-DQA1 [31]. The odds ratio for idiopathic MN with homozygosity for both risk alleles was 78.5. However, sequencing of all exons in the PLA2R1 gene failed to detect a causative allele that might affect the protein configuration [32]. Genotype-phenotype correlations have shown that those who carry a high-risk haplotype in the HLA–DQA1 and PLA2R1 genes have a much higher prevalence of circulating anti-PLA2R [33]. Kanigicherla et al. also showed higher anti-PLA2R titers in subjects carrying one or two alleles for HLA DQA1*05:01 or for DQB1*02:01 than in those with neither of these HLA alleles [34]. It is interesting that the “risk” alleles at the PLA2R1 locus are actually the major alleles, indicating that a large proportion of the population might be at risk for the disease. Some have proposed that the rare confluence of several relatively common factors: a particular isoform of HLA-DQA1 that confers susceptibility to autoimmunity, polymorphisms in PLA2R1 that alter expression and/or create a unique conformation identified by HLA class II on antigen-presenting cells, environmental factors, and perhaps age-related changes in immunoglobulin glycosylation, are all necessary for activation of the lectin pathway of complement and development of MN [27].
11.7 Do Anti-PLA2R Levels Relate to Disease Activity?
As many as 70–80 % of patients with primary MN have circulating anti-PLA2R antibodies, with no major ethnic differences in prevalence of these antibodies among diverse patient populations such as the US, European Caucasians, African-Americans, Chinese, and Koreans [34–38]. It is of interest that Japanese patients with primary MN may be somewhat unique as they have been shown to have a lower prevalence of PLA2R-associated MN [39]. Disease in this population has also been reported to be somewhat more benign and responsive to corticosteroid monotherapy, perhaps reflecting a genetic attenuation of disease severity.
Hofstra et al. were the first to demonstrate that circulating anti-PLA2R antibodies correlated strongly with clinical status [40]. In the 14 patients that were anti-PLA2R positive in the study, antibody levels correlated positively with proteinuria, serum β2-microglobulin, urinary IgG excretion, and serum creatinine at baseline. A strong correlation was also demonstrated with clinical status during follow-up, with anti-PLA2R levels decreasing significantly during remission and increasing again at relapse.
A subsequent study by Qin et al. showed that anti-PLA2R antibody levels correlate with development of remission on follow-up [36]. Remission occurred in 50 % of the patients with low titers of anti-PLA2R, and these patients developed no relapse. On the other hand, remission was observed in only 30 % of the patients with high titers of anti-PLA2R antibodies, and the average time to remission was more than double than that for patients with low antibody titers (14.47 ± 7.62 months versus 6.60 ± 3.58 months). Anti-PLA2R antibody testing in 21 additional idiopathic MN patients who had entered a clinical remission after treatment showed that anti-PLA2R was negative in 17 (81.0 %), while two of the patients with high titers of anti-PLA2R antibodies relapsed 6 months after entering a remission. This study showed that the presence of high-titer anti-PLA2R is associated with clinical disease activity and that a significant proportion of individuals become anti-PLA2R negative when they achieve clinical remission. However, patients with high anti-PLA2R antibody levels were prone to relapse.
In a subsequent study, Hofstra et al. extended their previous observations as well as pointed out that the rate of spontaneous remissions correlated with levels of anti-PLA2R [41]. Spontaneous remission occurred in 38 % and 31 % of the patients with low (41–175 U/ml) or moderate (176–610 U/ml) levels of anti-PLA2R by ELISA, while only one spontaneous remission occurred in the patients with anti-PLA2R titers in the highest tertile (>610 U/ml) [41]. Another group showed that PLA2R antibody levels fell over time in patients with spontaneous remission but remained elevated in patients who did not show a reduction in proteinuria [42].
Further evidence that anti-PLA2R level correlates with disease activity came from the work by Kanigicherla et al. [34]. Studying 90 prevalent patients with MN, these investigators found that 75 % of the patients with active disease were positive for anti-PLA2R antibodies. This contrasted with positivity in only 37 % of patients in partial remission and 10 % of patients in complete remission. Anti-PLA2R levels were significantly higher in patients with active disease compared to patients in partial or complete remission.
Anti-PLA2R levels can also predict patients initially presenting with sub-nephrotic-range proteinuria who are likely to progress to full nephrotic-range proteinuria. Hoxha et al. evaluated whether anti-PLA2R levels might predict development of nephrotic syndrome in patients with sub-nephrotic proteinuria under treatment with blockers of the renin-angiotensin system [43]. Significantly more anti-PLA2R-positive patients developed nephrotic-range proteinuria (13 out of 16) compared to anti-PLA2R negative patients (5 out of 17). At the same time, the anti-PLA2R-positive patients developed a more severe nephrotic syndrome compared to anti-PLA2R-negative patients. The use of immunosuppressive therapy was also more frequent in anti-PLA2R-positive patients (13 of 16 patients, 81 %) compared to anti-PLA2R-negative patients (2 of 17 patients, 12 %).
The overall available evidence shows that there is good correlation between the presence of circulating anti-PLA2R antibodies and the presence of active disease. As long as the autoantibody level remains above a certain threshold (which may vary from patient to patient), subepithelial deposits will continue to form, and the ongoing complement activation will keep the podocyte in an injured, “nephrotic” state. Even when anti-PLA2R disappears from the circulation, it may take months to years for the proteinuria to dissipate [44]. This is due to the fact that the changes in the glomerular basement membrane take a prolonged period of time to be repaired. Repeat biopsy studies in patients who have been successfully treated with the B-cell-depleting agent rituximab, or who have had serial biopsies after developing recurrent MN in the allograft, show the persistence of deposits containing IgG and complement components for a significant amount of time [24, 45]. Partial remission (proteinuria less than 3.5 g per day) is often achieved initially and may progress to a complete remission once the deposits have been resorbed and the podocyte has had sufficient time to reestablish its cytoskeletal structure and reformed slit diaphragms. Patients who are left with several grams of proteinuria likely have permanent structural damage in the form of secondary FSGS as well as tubular atrophy and fibrosis.
11.8 High Anti-PLA2R Levels Are Associated with an Increased Risk of Kidney Function Decline
In the study by Kanigicherla et al., high levels of anti-PLA2R antibodies correlated with degree of proteinuria and were associated with a high risk of declining kidney function over time [34]. Survival analysis showed that more than 50 % of the patients with high anti-PLA2R levels doubled the serum creatinine over a 5-year follow-up. Similarly, Hoxha et al. prospectively evaluated the role of anti-PLA2R antibodies on the increase of serum creatinine in 118 consecutive patients with MN [46]. Patients were divided into three groups according to their antibody levels at the time of inclusion in the study: low levels (20–86 RU/ml by a commercial ELISA), medium (87–201 RU/ml), and high (≥202 RU/ml). After a median follow-up time of 27 months (interquartile range 18–33 months), the clinical end point, defined as an increase of serum creatinine by ≥25 % and serum creatinine ≥ 1.3 mg/dl, was reached in 69 % of patients in the high antibodies level group versus 25 % of patients with low antibody levels. Patients in the high antibodies level group reached study end point faster (17.7 months) than in patients in the low group (30.9 months). Multivariate Cox regression analysis showed that anti-PLA2R levels were an independent predictor for progressive loss of kidney function. Although serum creatinine levels did not correlate with the degree of interstitial fibrosis, significantly more patients with moderate interstitial fibrosis were seen in the high anti-PLA2R group than in the low group.
11.9 Are Anti-PLA2R Levels Helpful to Guide Treatment in MN?
Beck et al. were the first to demonstrate that reduction or disappearance of anti-PLA2R antibodies following treatment with rituximab (RTX) was associated with clinical outcome [44]. These investigators quantified anti-PLA2R levels in serial serum samples from 35 patients with MN treated with RTX [44]. Baseline samples from 25 of 35 (71 %) patients were positive for anti-PLA2R, with autoantibodies declining or becoming negative in 17 (68 %) of these patients within 12 months after RTX. Patients who demonstrated such an immunologic response fared better clinically: 59 % and 88 % attained complete or partial remission by 12 and 24 months, respectively, compared with 0 % and 33 % among those who remained anti-PLA2R positive. Changes in antibody levels preceded changes in proteinuria (Fig. 11.3). Similarly, Hoxha studied five patients treated with RTX and found that anti-PLA2R antibody levels decreased in three patients that achieved partial remission after 12–18 months [35].
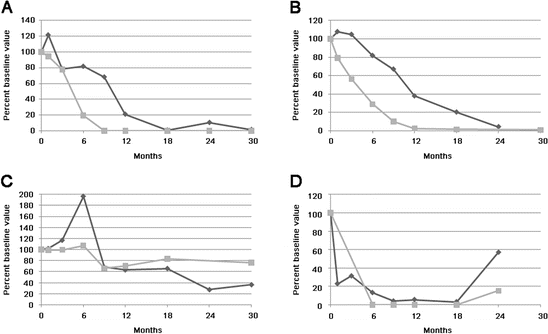
Fig. 11.3
Representative plots of anti-PLA2R (gray squares) and proteinuria (black diamonds) versus time following initial rituximab treatment. Values are plotted as percent of baseline value for the sake of better comparison between subjects (Reproduced from Ref. [44] with permission)
Most recently, Ruggenenti et al. reported on 132 MN patients treated with RTX [47]. Over a median follow-up of 30.8 months, 84 (64 %) patients achieved complete or partial remission of proteinuria. Lower anti-PLA2R antibody levels at baseline and full antibody depletion 6 months post-RTX were strong predictors of remission. All 25 complete remissions were preceded by complete anti-PLA2R antibody depletion. Reemergence of circulating antibodies predicted disease relapse.
The ability of monitoring anti-PLA2R levels to predict clinical outcome appears independent of the type of immunosuppressive agent used. Bech et al. studied 48 patients treated with oral cyclophosphamide (CYC) or mycophenolate mofetil (MMF) in combination with corticosteroids for 12 months [48]. At baseline, 71 % of the patients were positive for anti-PLA2R. Immunosuppressive treatment resulted in a rapid decrease of antibody levels with antibody status at the end of therapy predicting long-term outcome. With follow-up, 58 % of the patients that became anti-PLA2R negative were in remission compared with 0 % of antibody-positive patients. Baseline antibody levels did not predict response to therapy. Both MMF and CYC resulted in a rapid decrease of antibodies, but patients treated with CYC became anti-PLA2R negative faster than patients treated with MMF, with significantly more patients treated with CYC becoming anti-PLA2R negative (16 of 18 for CYC versus 8 of 15 for MMF) at the end of therapy. As discussed by Glassock in an accompanying editorial, levels of anti-PLA2R at the end of treatment predicted the long-term outcomes: 55 % of those who were antibody negative at the end of therapy remained in remission for 5 years compared to none in remission at 2 years following completion of treatment in the anti-PLA2R-positive group [49].
Similar results were also found for patients treated with calcineurin inhibitors. Hoxha et al. evaluated response to immunosuppressive therapy in 101 patients with MN and positive anti-PLA2R antibodies [42]. Fifty-four patients were treated with a calcineurin inhibitor, 35 with an alkylating agent, and 9 with RTX. Immunosuppressive therapy led to a sustained 81 % reduction in anti-PLA2R antibody levels paralleled by a 39 % reduction in proteinuria. However, the investigators could not find statistically significant differences in response between the patients receiving a calcineurin inhibitor or other treatment agent. The study also showed that the use of immunosuppressive therapy led to reduction in anti-PLA2R antibody levels that parallels reduction in proteinuria and confirmed previous observations for a time lag between reduction in anti-PLA2R levels and remission of proteinuria [35, 44]. Most recently, Hladunewich et al. also reported that clearing of serum anti-PLA2R antibodies prior to or in parallel with improvement in proteinuria was noted in some, but not all patients treated with natural ACTH (H.P. Acthar® Gel) [50].
These observations strongly suggest that serial measurement of anti-PLA2R antibody levels may help in monitoring disease activity and can predict the response to immunosuppressive therapy. As such, quantification of antibody levels appears as more specific and an earlier way of determining response to therapy as compared with the clinical response of proteinuria and a means to individually tailor therapy.
11.10 Assays for Anti-PLA2R
Although initial studies on the association of MN and anti-PLA2R utilized a Western blot assay performed in the research setting, subsequent studies have capitalized on the availability of several commercially available assays that are now available for clinical use [51, 52]. An indirect immunofluorescence test provides semiquantitative titers by assaying serial dilutions of patient serum against biochips containing cells transfected with human PLA2R or untransfected control cells. A higher-throughput ELISA for anti-PLA2R is also available that provides a quantitative titer based on reference standards. It should be noted that the values reported based on this commercial ELISA vary in range from those reported from several publications using an in-house, research ELISA for anti-PLA2R. The threshold for seropositivity for the commercial assay was chosen to maximize specificity but may falsely label certain low-titer samples as negative. The use of a lower threshold (e.g., 2 RU/ml) may more accurately detect low-titer samples without sacrificing specificity [53].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 History of Research on Pathogenesis of Idiopathic Nephrotic Syndrome
Regulatory T Cells and Oxidative Stress in Minimal Change Nephropathy
Cytokines as Active Factors in Minimal Change Nephrotic Syndrome
Podocytes as a Direct Target of Drugs Used in Idiopathic Nephrotic Syndrome
Co-stimulatory Molecule CD80 (B7.1) in MCNS
History of Research on Pathogenesis of Idiopathic Nephrotic Syndrome
Regulatory T Cells and Oxidative Stress in Minimal Change Nephropathy
Cytokines as Active Factors in Minimal Change Nephrotic Syndrome
Podocytes as a Direct Target of Drugs Used in Idiopathic Nephrotic Syndrome
Co-stimulatory Molecule CD80 (B7.1) in MCNS
 Cationic Bovine Serum Albumin as Cause of Membranous Nephropathy: From Mice to Men
Cationic Bovine Serum Albumin as Cause of Membranous Nephropathy: From Mice to Men
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


