and C viruses.17, 18, 19, 20 Of these, EBV has already been a subject of interest for many years.
Table 4-1 Tumor Site of Gastrointestinal Lymphomas | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
is important for most extranodal tumors, including many GI lymphomas. This REAL classification was upgraded in 1999 and has now been adopted by the WHO classification.27, 28, 29 While the lymphoma classifications will continue to evolve, disease-specific rather than morphology-centered classification is likely to remain a central theme.
Table 4-2 Classification of GI Lymphomas | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
related fields, GI lymphoma is increasingly recognized at an early stage. Particularly with regard to gastric lymphoma, presentation with a large mass lesion is a rarity these days in developed countries. With the increasing use of video capsule endoscopy, one could expect similar changes with regard to intestinal lymphomas. As increasingly early lesions are diagnosed, the role of the pathologist has shifted from merely subtyping or grading the lymphoma or both to differentiating it from benign and reactive disorders. It should also be recognized that traditional staging systems are poor in predicting the prognosis of primary GI lymphomas, particularly MALT lymphomas. Alternate staging systems have been developed, of which modified Ann-Arbor system is most widely used in practice (Table 4-3).30 Distinguishing benign from malignant lymphoproliferative lesions of the gut can be difficult due to small biopsy size, tissue artifacts, limited tissue to assess the architectural pattern, confounding presence of lymphoid follicles that can be seen in both benign and malignant lesions, or because lymphoid markers may not be either readily available or easy to carry out in biopsies. In such situations, application of advanced molecular diagnostics could be of immense help.
Table 4-3 Modified Ann-Arbor Staging System for Primary Gastrointestinal Lymphomas | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
conditions. Using sensitive techniques, about 10% to 15% cases of H. pylori-related chronic gastritis without any histologic evidence of lymphoma may show clonal B-cell populations. The choice of the molecular technique in a given situation depends upon the clinical setting, target to be assayed and the test availability. Further discussion of the technical aspects of molecular tests and their limitations are beyond the scope of this book.
outcome, in routine practice, presence of clonal B-cell proliferation in absence of histologic evidence of lymphoma cannot be totally disregarded.39
where it is a cause of nodularity; when appendiceal lymphoid tissue can be seen during endoscopic screening of mucosa at the appendiceal orifice and is biopsied as a “mucosal polyp”; in resected appendices, mostly for acute appendicitis, when obliteration of its lumen could be used as indirect evidence of lymphoid hyperplasia; in children in whom it may form the apex of an intussusception, often associated with adenovirus infection, and in diversion colitis/proctitis when the lymphoid hyperplasia can be seen grossly. Histologically, it is even more difficult to define lymphoid hyperplasia. The presence of lymphoid follicles with large secondary germinal center often correlates with grossly enlarged lymphoid nodules. The challenge is often to ensure that there are plasma cells present and that this is not a manifestation of common variable immunodeficiency (CVID) disease, and, if it is, that organisms such as Giardia are not present.
admixed with lymphoid follicles with germinal centers and fibrosis (Fig. 4-2). Since reactive lymphoid follicles may also be found within gastric lymphomas, one cannot rely on these follicles for distinguishing lymphoid hyperplasia from lymphoma. Clumps of IELs mimicking lymphoepithelial lesions of MALT lymphomas may also occasionally be found, but they differ from the latter in that they consist of mature lymphocytes, mostly T cells, usually have less than three cells, and do not cause epithelial destruction or degeneration.
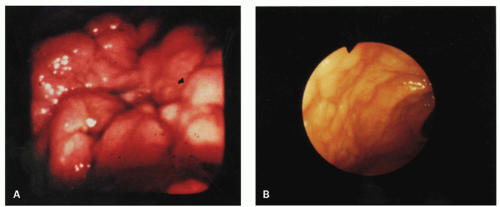 Figure 4-1. Endoscopic appearances of gastric lymphoid hyperplasia. A: Central ulceration surrounded by severe mucosal nodularity. B: Plaque-like thickening of rugal folds without ulceration. |
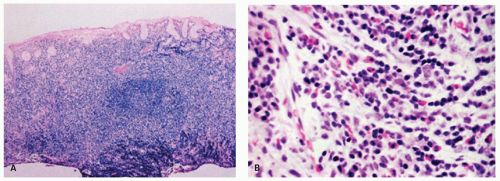 Figure 4-2. Endoscopic biopsy specimen of gastric lymphoid hyperplasia. A: Low-power view showing a dense mucosal inflammatory infiltrate. B: Detail of part (A) showing a polymorphous infiltrate composed of mature lymphocytes, plasma cells, eosinophils, and histiocytes. |
differential diagnosis of MALT lymphoma).59 Presence of a clonally rearranged Ig gene detected by PCR in this setting is likely to represent a lymphoma, and the histology needs to be interpreted with extreme caution.59, 60
in the literature that were clearly distinguished from a lymphoma.49, 70 Patients usually present with recurrent abdominal pain. Radiologic examination may show a focal constricting lesion. Grossly, there is nodular thickening of the mucosa, which may be circumferential (Fig. 4-7). Microscopically, the lymphoid infiltrate may involve only the mucosa and submucosa or, less frequently, the full thickness of the intestinal wall. The differential diagnosis is not a problem because of the mature, bland nature of the lymphoid infiltrate and the presence of follicles with germinal centers throughout the lesion.49 The lamina propria should be examined to ensure that adequate numbers of plasma cells are present, and that this is not secondary to a B-cell lymphoma. If there is any doubt, immunohistochemistry can be performed to ensure particularly that most of the plasma cells are IgA and that this is not part of IgA deficiency or CVID syndrome. Confirmation can be obtained from serum immunoglobulins.
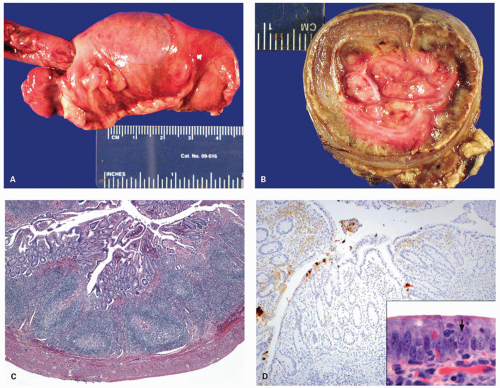 Figure 4-3. Lymphoid hyperplasia in small bowel and colon leading to intussusception of terminal ileum into the colon (A). The transverse section showing fleshy tissue in the invaginated segment representing lymphoid hyperplasia (B). Histology of the lymphoid hyperplasia in the colonic segment from the same case showing numerous lymphoid follicles with reactive germinal centers (C). The immunostain for adenovirus shows strong positivity in the overlying epithelium (D). Note the adenovirus nuclear inclusions (arrow) in the overlying epithelium (inset). |
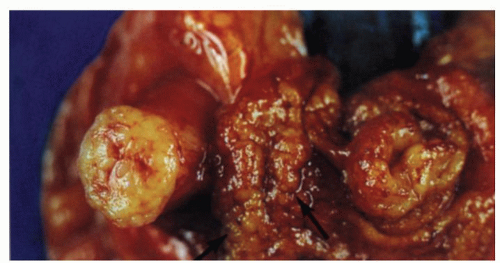 Figure 4-4. Focal lymphoid hyperplasia of the terminal ileum and appendix from a patient with symptoms of acute appendicitis. Gross appearances showing cobble stoning of ileal mucosa (arrow) and marked thickening of the appendix with luminal obliteration. |
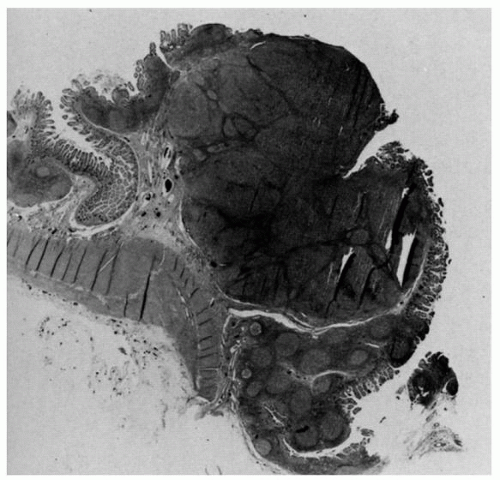 Figure 4-5. Focal lymphoid hyperplasia of the terminal ileum and appendix. Note the follicular hyperplasia of Peyer’s patch in the terminal ileum, on the right, extending into the appendix. |
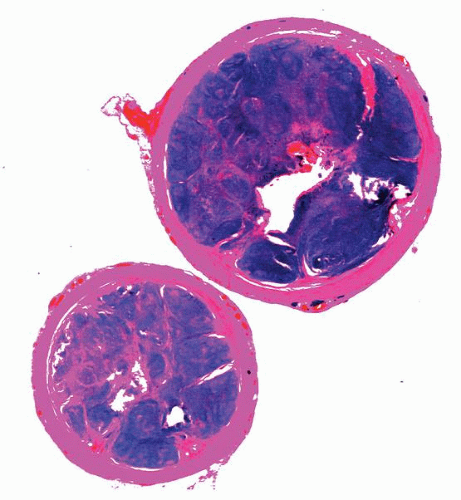 Figure 4-6. Focal lymphoid hyperplasia of the appendix. Lower-power photomicrograph of a cross-sectional cut demonstrating marked mucosal thickening, follicular hyperplasia, and luminal obliteration. |
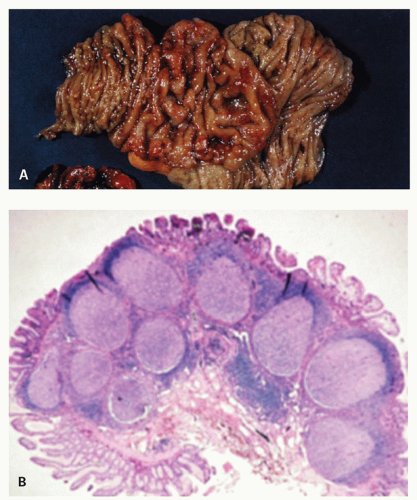 Figure 4-7. Focal lymphoid hyperplasia of the duodenum. A: The mucosal folds are focally thickened and edematous. Additionally, there are numerous nodules in both the edematous and nonedematous mucosa. B: Low-power photomicrograph of part (A) illustrating hyperplastic lymphoid follicles in the mucosa and submucosa. (Courtesy M. Ranchod, M.D.) |
absent. Focal lymphoid hyperplasia of the rectum is readily differentiated from malignant lymphoma (see the discussion of lymphoid hyperplasia of terminal ileum). It should also be kept in mind that malignant lymphoma is exceedingly rare at this site.13 As discussed in the preceding section, an immunodeficiency syndrome should be excluded.
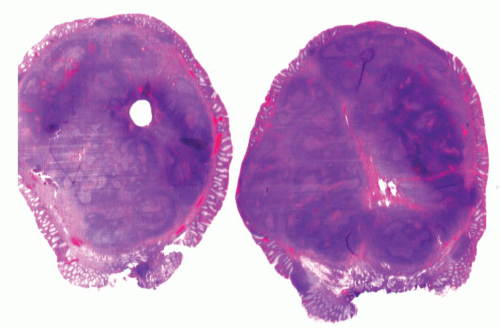 Figure 4-8. Focal lymphoid hyperplasia of the rectum (lymphoid polyp) demonstrating the polypoid appearance of the lesion. The lesion is covered by intact, focally attenuated colonic epithelium and contains large lymphoid follicles with prominent germinal centers. |
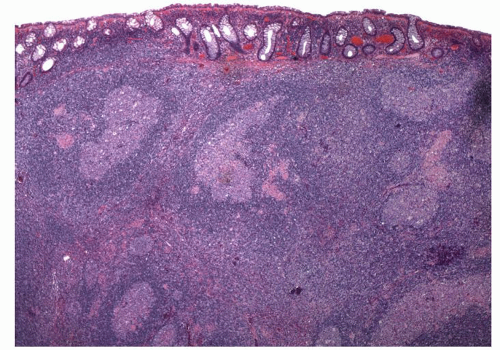 Figure 4-9. Microscopic appearance of rectal lymphoid hyperplasia showing numerous lymphoid follicles with germinal centers. Note that the infiltrate tends to push the crypts apart. |
immunodeficiency syndromes (see also Chapter 5).
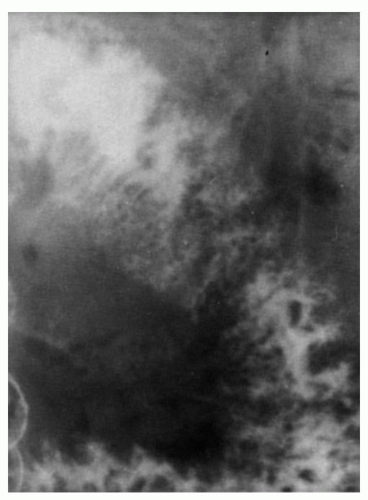 Figure 4-10. Nodular lymphoid hyperplasia with hypogammaglobulinemia: x-ray appearances. The barium radiograph is characterized by numerous nodular translucencies representing the mucosal lymphoid nodules. |
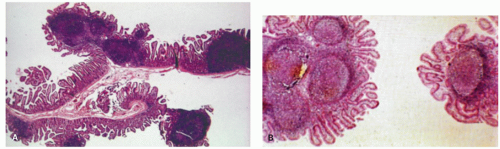 Figure 4-11. Low-power view showing hyperplastic mucosal lymphoid follicles. The mucosal nodules are composed of either (A) single or (B) clusters of hyperplastic lymphoid nodules. |
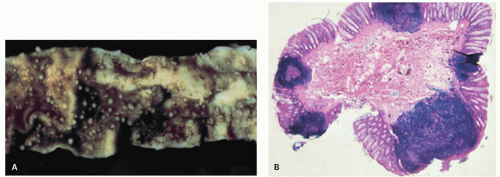 Figure 4-12. Nodular lymphoid hyperplasia of the large intestine. A: Showing mucosa studded with small, sessile nodules. B: Showing hyperplastic mucosal lymphoid follicles. |
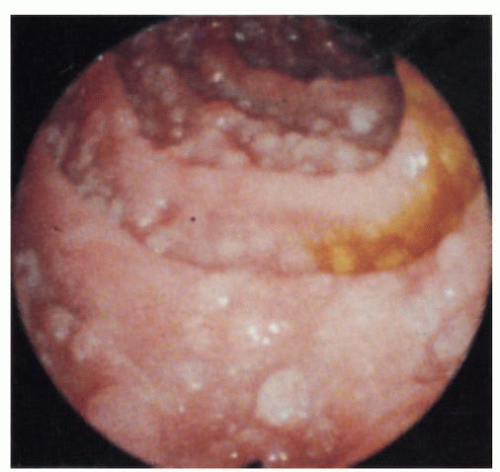 Figure 4-13. Nodular lymphoid hyperplasia of the large intestine: endoscopic appearance. The mucosa is studded with numerous pearly white nodules. (Courtesy S. Weiss, M.D.) |
surrounded by a diffuse infiltrate of CLC and a variable degree of plasma cell differentiation. Tumor cells infiltrating the overlying squamous epithelium are often present, suggesting epitheliotropism similar to lymphoepithelial lesions of gastric MALT lymphomas. Due to rarity of these lesions, it is unknown if the molecular genetics of these tumors is similar to MALT lymphomas at other sites. DLBCLs similar to their nodal counterparts are also known to arise in esophagus. Primary esophageal lymphomas including T-cell lymphoma, large-cell anaplastic Ki-1 lymphoma, Hodgkin’s lymphoma, and plasmacytoma have been reported in the literature and are exceedingly rare.97, 98, 106, 110, 112, 113
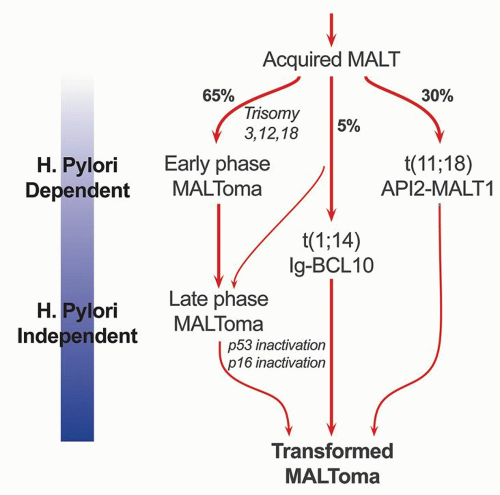 Figure 4-14. Schematic diagram showing the progression of H. pylori-driven B-cell proliferation leading to lymphoma. |
and that H. pylori is the most likely antigen. It is also clear from these experiments that T cells play a vital role in the B-cell proliferation and that the B cells themselves are not H. pylori specific but make antibodies to a variety of autoantigens, including parietal cells and the proton pump that can result in gradual atrophy of specialized gastric mucosa. While the initial B-cell proliferation is H. pylori driven and polycloncal, subsequent molecular and genetic alterations lead to clonal expansion and autonomous growth. Whether H. pylori-specific factors play a role in the development of lymphoma is still somewhat controversial.127, 128, 129 The H. pylori strains, which accelerate the tumor growth, include the Cag A strain, a gene found in approximately 60% of H. pylori organisms, and the vac A (vacuolating cytotoxin) genotype.129, 130, 131 MALT lymphomas associated with H. heilmanni infection have also been reported.132 The cell of origin of MALT lymphomas is a B-cell that is identical to lymphocytes of the marginal zone of lymphoid follicles. These represent postfollicular B cells, and antigen selection occurs in some cases.133, 134, 135
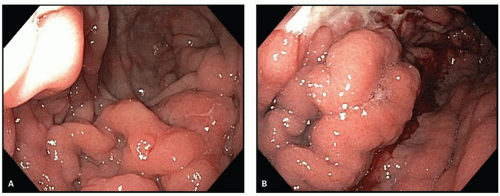 Figure 4-15. Lymphoma of the stomach: endoscopic appearances. A: Showing nodular mucosal lesions. B: Showing irregular mucosal masses, which may be indistinguishable from carcinoma. (Courtesy J. Nord, M.D.) |
although many foci may be microscopic in size. Once the infiltrate extends into the submucosa, it tends to separate the muscle fibers of the muscularis mucosae and either grows in a band-like manner, with a pushing border, or invades with an irregularly infiltrating margin. Eventually, the muscle undergoes atrophy, resulting in the marked weakening of the viscus and a propensity to perforation. The regional lymph node involvement is distinctly uncommon with superficial lesions and is seen once the tumor spreads to the muscularis propria or undergoes transformation to high grade.
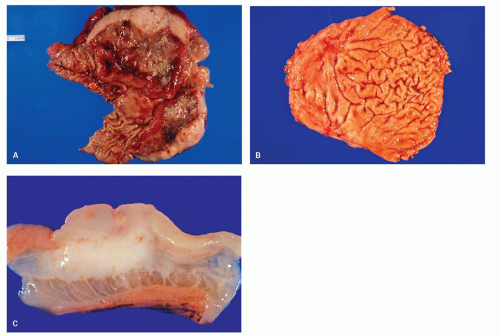 Figure 4-16. A: Gross photograph of a resection specimen of stomach showing large ulcerating lesion with marked thickening of the stomach wall. The cut section shows fleshy grey-white tumor diffusely involving the wall without any well-defined borders. B: Another specimen of stomach showing smaller ulcerating lesion with thickening of the mucosal folds around it. C: The cut section shows fleshy grey-white tumor infiltrating into the submucosa and muscularis propria without any well-defined borders. |
tend to spread out from the marginal zone of the follicle to infiltrate the interfollicular zone and destroy the glands. Occasionally, these cells may also infiltrate the center of the follicle (follicular colonization), producing a vague nodular architecture (Fig. 4-18).141 This can be easily demonstrated by dendritic cell markers (CD21, CD23, or CD35) that highlight the background of dendritic reticulum cells in the colonized follicle. Another invariable finding in over 90% of these patients is the presence of H. pylori infection.
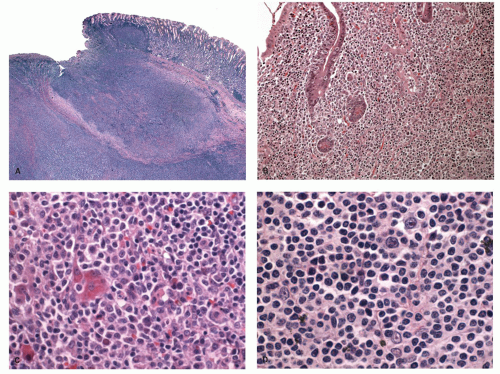 Figure 4-17. Microscopic appearance of gastric malt lymphoma. A: Low power showing extensive lymphocytic infiltrate with ulceration of mucosa and extension into the wall of the stomach. B: Lymphoepithelial lesions showing clusters of monocytoid B-cell infiltrating the glands. C: High power showing mixture of cells with many small lymphocyte-like cells, centrocyte-like cells, and plasma cells. D: High power showing large centroblasts/immunoblast-like cells interspersed among small lymphocyte-like cells. |
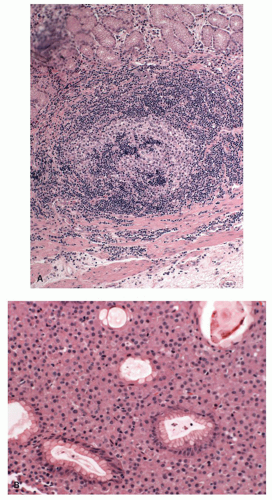 Figure 4-18. A: Microscopic appearance of follicular invasion by the marginal bone cells in a case of malt lymphoma of the stomach. B: Malt lymphoma of the stomach showing a predominant population of plasma cells. Often the plasma cells are localized in the superficial lamina propria. |
genes.155, 156, 157 The API2 is an apoptosis inhibitor, whereas MALT1 is involved in antigen receptor-mediated NFkB activation. The fusion transcript is consistently expressed in these cases and is believed to be oncogenic. Unlike wild-type API2 and MALT1, API2-MALT1 fusion product is a potent activator of NFkB. NFkB activation in turn leads to activation of other cytokine and growth factor genes that are important for cellular activation, proliferation, and survival, thus contributing to tumor development.158, 159, 160 This translocation is seen in about 15% to 30% of gastric MALT lymphomas.161 The same translocation has also been described in MALT lymphomas of other sites including skin and lungs. However, it is absent in nodal and splenic marginal zone lymphomas.162, 163, 164, 165 Cases of H. pylori gastritis also do not show this translocation. MALT lymphomas with this translocation do not respond to H. pylori eradication and more often spread to distant sites including regional lymph nodes.166 An initial report suggested that despite the association of t(11;18)(q21;q21) with adverse clinical features, the translocation is uncommonly found in cases with DLBCLs or so-called transformed MALT lymphomas, that is, low-grade MALT lymphomas with a large-cell component.167, 168 A subsequent study however shows that this translocation is seen with almost equal frequency in transformed MALT lymphomas or DLBCLs of the stomach.169 Even if found, H. pylori eradication treatment is undertaken together with other lymphoma therapy.
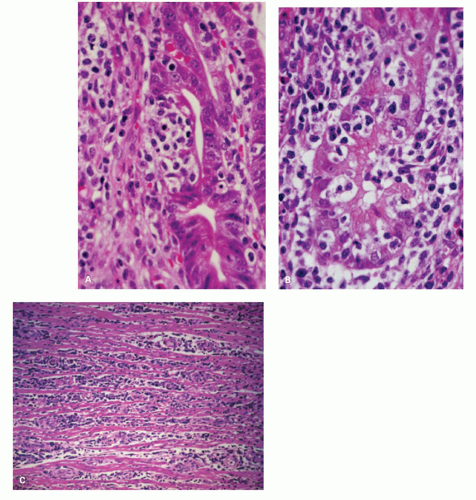 Figure 4-19. MALT lymphoma of the stomach. A,B: Medium-power photomicrographs showing infiltration of glands by the atypical lymphocytes. C: Lymphoma showing separation of the muscle fibers of the muscularis propria by the lymphomatous infiltrate. |
associated with trisomies 3, 12, and 18, unlike t(11;18) (q21;q21).146 The translocation brings the entire BCL10 gene under the regulatory control of the Ig gene and deregulates its expression.143, 170 Earlier studies showed that although BCL10 activates NFkB, it is weakly proapoptotic.171, 172 However, subsequent studies have shown that BCL10 does not have proapoptotic activity in vivo.173, 174 It is essential for the development of B- and T-cell functions and specifically links the antigen receptor signaling to NFkB pathway.175, 176, 177 In response to the surface antigen receptor stimulation, BCL10 oligomerizes and binds to MALT1, leading to MALT1 oligomerizartion that in turn activates NFkB. In line with the physiological role of BCL10 in normal B cells, the protein is expressed predominantly in the cytoplasm of germinal center B cells.178 BCL10 is strongly expressed in the nuclei of MALT lymphomas with t(1;14)(p22;q32) or its variants.178 However, moderate nuclear BCL10 expression is also seen in about 50% of cases negative for this translocation and in almost all t(11;18)(q21;q21)-positive cases.161, 165, 179 Studies using BCL10 immunostains and interphase FISH analysis show that t(1;14)(p22;q32) is specifically associated with MALT lymphomas and not with other lymphoma subtypes.165 This translocation occurs in about 5% of gastric MALT lymphomas and has also been associated with MALT lymphomas of lungs.165 Gastric MALT lymphomas with this translocation are typically at advanced stages and unlikely to respond to H. pylori eradication.
lymphomas in biopsies. When the biopsies are adequate and the morphology and immunohistochemistry are typical, many are comfortable making the diagnosis, while some always hedge by stating “consistent with”. One of the problems with MALT lymphomas is the lack of a distinct immunohistochemical profile, and stains are largely used to exclude other lymphoma types (mantle, follicular, CLL etc). Presence of one of the MALT associated translocation should certainly help one in making a definitive diagnosis however, these are present only in a small proportion of cases and are still not readily available in all labs. Nevertheless, pathologists should still be comfortable making this diagnosis in the presence of appropriate histology with the knowledge that the first line of therapy is Helicobacter eradication followed by re-endoscopy and biopsy a few months later. If the morphology and immunohistochemistry are all correct, but Helicobacter cannot be demonstrated or the biopsies are too small, reporting them as “atypical lymphoid infiltrate” and suggest follow-up biopsies for a full lymphoma work-up with flow-cytometry, molecular assays and serology for Helicobacter.
extremely valuable in this setting. However, if the sample is small and the findings are inconclusive, additional biopsies for diagnostic workup should be requested. CLL/SLLs have often involvement of peripheral blood, and knowledge of the peripheral blood findings could be helpful.
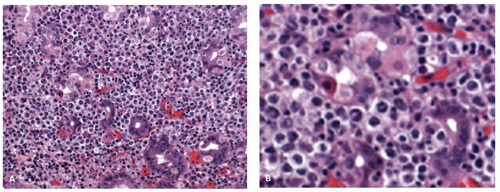 Figure 4-20. A: Appearance of DLBCL of the stomach. The morphology resembles that of an extranodal DLBCL of the stomach. B: There are numerous large cells in a background of H. pylori-positive MALT-type lymphomas and lymphoepithelial lesions are still clearly visible. |
Related posts:
 Gastrointestinal Manifestations of Extraintestinal Disorders and Systemic Disease
Gastrointestinal Manifestations of Extraintestinal Disorders and Systemic Disease
 Esophagus: Normal Structures, Developmental Abnormalities, and Miscellaneous Disorders
Esophagus: Normal Structures, Developmental Abnormalities, and Miscellaneous Disorders
 Inflammatory Disorders of the Esophagus: Reflux and Nonreflux Types
Inflammatory Disorders of the Esophagus: Reflux and Nonreflux Types
 Small Bowel Mucosal Disease
Small Bowel Mucosal Disease
 Stomach and Proximal Duodenum: Inflammatory and Miscellaneous Disorders
Stomach and Proximal Duodenum: Inflammatory and Miscellaneous Disorders
 Small and Large Bowel Polyps and Tumors
Small and Large Bowel Polyps and Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


