Diameter (cm)
Metastatic potential (%)
<1.0
3–5
1.0–1.9
10–30
≥2.0
>75
The majority of diagnosed rectal carcinoids (65–80 %) will be less than 1 cm in diameter, which carries a 3–5 % risk of metastasis [5, 10, 33]. Both local transanal excision and endoscopic resection has demonstrated to be safe and curative for the vast majority of patients with these small carcinoids that lack adverse features – tumors less than 10 mm, without invasion of the muscularis propria and without ulceration, or less than 10 mm with adequate endoscopic surveillance [19, 36]. Rectal carcinoids ranging from 1 to 1.9 cm in diameter are associated with a 10–30 % chance of metastases [5]. Transanal excision is commonly performed for intermediate-sized rectal lesions, confined to the submucosa, without histologic risk factors [16, 37]. Most authorities recommend that patients with tumors of 1–1.9 cm in size, with invasion of the muscularis propria or other adverse features, and no evidence of metastatic disease, should undergo low anterior resection (LAR) or abdominoperineal resection (APR) with mesorectal excision [11, 17]. Lesions ≥2 cm have a 75 % chance of metastasizing [37], a median survival of 7 months [8], and a 10-year mortality of 60 %. Most studies support the use of APR or LAR with mesorectal excision for treatment of these larger tumors both for cure and for palliation [4, 16, 36, 37]. Aggressive surgical intervention, however, has never been shown to improve survival compared with local excision in tumors >2 cm. Adjuvant chemotherapy and radiotherapy has occasionally been used for large carcinoids, without clear evidence of benefit [17]. Our algorithm for the treatment of rectal carcinoids based on size is depicted in Fig. 22.1.
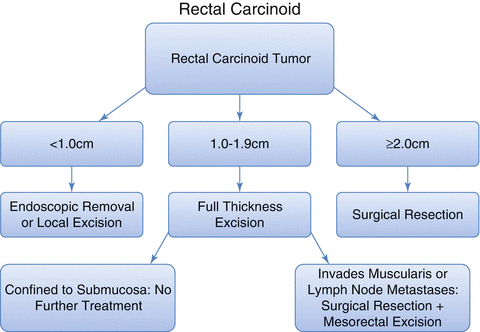
Fig. 22.1
Suggested algorithm for management of rectal carcinoid based upon tumor size
The most frequent sites of rectal carcinoid metastasis are lymph nodes and liver, with less common sites including brain, bone, peritoneum and lung [5, 8, 9]. Various combinations of chemotherapeutic agents have been used for metastatic rectal carcinoids, with little improvement in survival outcomes [8]. Lastly, neuroendocrine tumors can recur many years after surgical resection. The vast majority of carcinoids are Stage I tumors, or submucosal tumors less than 2 cm in size, and have an extremely low risk of recurrence. Therefore, there is no clear indication to perform long-term endoscopic or radiographic surveillance. Routine annual radiographic surveillance with CT or MRI should be considered for Stage II (invading muscularis) or III (regional lymph node involvement) NETs because of their high risk of systemic metastases, even years after treatment [16].
Lymphoma of the Rectum
Primary Lymphoma of the Rectum
The large intestine is the site of 6–12 % of all gastrointestinal lymphomas [38–40]. Rectal lymphoma can represent a primary rectal malignancy or metastases from a nodal origin, known as secondary rectal lymphoma. The vast majority of rectal lymphomas are sequelae of systemic lymphoma. Primary rectal lymphoma is exceedingly rare, representing only 0.2–0.4 % of all primary colorectal malignant neoplasms [39, 41–43]. Almost all primary colorectal lymphomas are non-Hodgkin, B cell lymphomas [42, 44]. Very few cases of Hodgkin lymphoma have been reported in the literature and these are often associated with HIV or EBV-infection, inflammatory bowel disease or immunocompromised hosts [45–47].
Differentiation of primary from secondary colorectal lymphoma is necessary because both the survival outcomes and the therapeutic management of the two are distinct. In 1961 Dawson et al. introduced criteria to diagnosis primary gastrointestinal lymphoma [48]. In order to classify a GI malignancy as a primary lymphoma several criteria must be met. These include: no palpable peripheral lymphadenopathy, no mediastinal lymphadenopathy, normal white blood cell count on peripheral blood smear, only mesenteric lymph nodes adjacent to the tumor are involved at laparotomy, and no malignant lymphomatous disease of the liver and spleen. One detriment of this classification system is the difficulty distinguishing between secondary lymphoma and those with primary GI lymphoma who present with widely metastatic disease. There is no standardized classification or staging system for GI lymphoma and lymphoma staging systems, designed for traditional nodal-based lymphoma, fail to provide adequate prognostic treatment guidance for primary GI lesions. Several staging systems exist for GI lymphoma including the Ann Arbor staging with Musshoff modification [49], the international prognostic index (IPI) [50], the Paris staging system [51] and the WHO classification system [52]. The Ann Arbor staging system, originally designed for Hodgkin’s lymphoma, and its Musshoff modification, adopted for extranodal disease in 1977, is a very elementary system. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project developed of the International Prognostic Index (IPI) for patients with a diffuse large B-cell lymphoma (DLBCL) that consisted of Ann Arbor stage, patient characteristics and simple laboratory measurements. The Paris staging system has increasingly gained significance due to its ability to distinguish primary from distant lymphoma manifestations depending on involved organ. Recently there has been greater advocacy for use of the WHO system, which characterizes lymphomas on the basis of morphology, immunophenotype, molecular genetics, and clinical features [53–55]. This system has allowed clinicians to better predict the clinical behavior of the lymphoma as well as modify management to achieve greater therapeutic success.
Clinical Presentation and Diagnosis
Primary rectal lymphomas are most often diagnosed in males in their fifth through seventh decade of life [56, 57]. Presenting symptoms, similar to other rectal neoplasms, include abdominal pain, weight loss, palpable abdominal mass or, most commonly, lower gastrointestinal bleeding [42, 58, 59]. Obstruction and perforation however are rare in patients with colorectal lymphoma [60]. Distal rectal lesions may be appreciated on physical examination; however, higher more proximal lesions require contrast enema or colonoscopy for identification. There are no unique radiologic or colonoscopic features to differentiate primary rectal lymphoma reliably from more common rectal tumors. Endoscopic appearance of colorectal lymphomas are variable and have been described as fungating, ulcerative, infiltrative, ulcerofungating, and ulceroinfiltrative types, with fungating and ulcerofungating types being more common [61]. CT scan and double-contrast barium enema demonstrate both focal and diffuse lesions [62]. Histologic patterns are variable and multiple biopsies with immunohistochemistry and molecular studies are often necessary for definitive diagnosis [57, 63].
Primary rectal lymphoma has been associated with a number of conditions, including longstanding ulcerative colitis [40, 64, 65], pelvic irradiation [66], HIV and EBV infections [67, 68], as well as solid organ transplantation [69]. A number of investigators have recently described primary rectal lymphomas arising in homosexual men [70–72]. While no definitive causal relationship has been identified, all of these conditions are known to cause immune system alterations, which may explain their association with primary rectal lymphoma.
Treatment and Prognosis
Given the rarity of primary colorectal lymphoma, studies concerning management are limited to small, retrospective observational studies. Treatment remains variable, however, historically it has been managed with surgical resection with or without the addition of chemotherapy. Depending on the stage of the lesion, surgical resection is performed both with curative intent and to prevent further complications such as hemorrhage, perforation or obstruction [42, 73, 74]. Surgical resection for primary rectal lymphoma includes LAR, APR, or transanal excision depending on the tumor location and extent of invasion. The role of radical surgery in the management of indolent rectal lymphoma is somewhat controversial due to the associated morbidity of rectal resection with a complication rate around 20 % [60, 75, 76]. Chemotherapy which includes cyclophosphamide, vincristine, doxorubicin and prednisolone (CHOP) is given to improve survival; using this regimen, disease-free survival rates of 35–45 % at 4 years have been realized in patients with aggressive lymphoma [77]. The addition of rituximab to standard CHOP-based therapy has also been showed to further improve survival outcomes [78]. Primary radiotherapy for unresectable lesions, high-risk surgical candidates, and patients who are unwilling to undergo surgery, have also been described, with some success [79–81]. Most authors advocate adjuvant radiotherapy or chemoradiation [43, 56, 75, 79].
Most authors advocate for adjuvant chemotherapy in addition to surgical resection. Avilés et al. demonstrated 10-year survival outcomes of 83 % in 53 patients with primary colonic lymphoma treated with complete surgical resection followed by chemotherapy. However, this is likely to be related to the very select population of Stage IE tumors, which were studied [75]. Overall 5-year survival for primary colorectal lymphoma is 30–60 % [56, 73, 79, 82–84]. As expected there is better survival for patients presenting with localized disease (50 % 5-year survival) compared with those presenting with regional lymph node metastasis (24 % 5-year survival) [79]. The histologic grade of the of primary lymphoma in addition to stage may also impact prognosis [43]. A management outline of rectal lymphoma is depicted in Fig. 22.2.
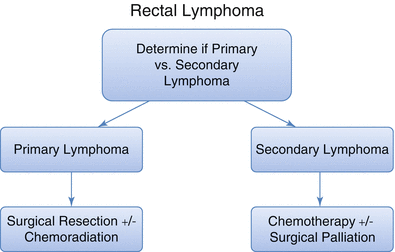
Fig. 22.2
Suggested algorithm for management of primary and secondary rectal lymphoma
Secondary Lymphoma of the Rectum
Secondary lymphoma of the rectum is defined as regional lymph node metastasis to the rectum. In patients with metastatic lymphoma of nodal origin, 5–46 % will have some degree of GI tract involvement [38, 79]. Presenting symptoms of metastatic lymphoma to the rectum are hematochezia and weight loss [56]. Primary therapy for metastatic lymphoma remains chemotherapy. Surgical intervention is generally of little benefit for the treatment of secondary rectal lymphoma unless it is for complications of these lesions such as intussusception, perforation, obstruction or uncontrollable hemorrhage. Lastly, prognosis is poor with overall 5-year survival of only 15 % [56].
Anorectal Melanoma
Anorectal melanoma (ARM), a type of mucosal melanoma, is an aggressive disease with a bleak prognosis. Although melanocytes do not normally occur in the rectal mucosa, malignant melanomas have been found to arise from areas of normal-appearing melanocytes, suggesting that certain individuals may have melanocytes present in the mucosa of the rectum [85, 86]. ARM is a rare tumor of the rectum, accounting for between 1 and 2 % of lower gastrointestinal malignancies and less than 2 % of all melanomas [87–89]. The incidence of ARM is thought to be increasing for reasons that are unclear [90–92]. After the head and neck and female genital tract, the anorectum is the third most common mucosal site of melanoma involvement [90].
Clinical Presentation and Diagnosis
ARM is commonly diagnosed in the sixth and seventh decades of life [93–96]. Anal bleeding is the most common symptom in those diagnosed with ARM [93, 94, 96]. The mean duration of symptoms before presentation is 5–6 months [93, 94, 96]. Multiple factors are thought to contribute to a delay in diagnosis of ARM. Frequently these lesions can be amelanotic in up to 20–25 % of cases; additionally, they are often confused with benign diseases such as hemorrhoids due to similar presenting symptomatology [88, 91, 94, 95, 97]. Two thirds of lesions are found in the anal canal or anal verge and approximately a third of cases are found in the distal rectum [92, 93]. While anal melanoma may vary slightly from rectal melanoma in initial presentation or recurrence patterns, recent data suggests there is no difference in survival outcomes by melanoma location within the anorectum [98].
Treatment and Prognosis
Despite multiple therapeutic approaches including surgical resection, radiation, and systemic therapy alone or in combination, prognosis and survival remain dismal for ARM [95, 98, 99]. More radical surgical resection has not been shown to alter survival outcomes. Even when patients are appropriately pathologically staged after rectal resection there is no survival difference between patients who undergo APR or wide local excision (WLE). There is a higher morbidity associated with APR; however, WLE may be associated with a higher rate of local recurrence [89, 95, 97, 100, 101]. There may be some role for radiation as palliative therapy in locally advanced, recurrent, or metastatic disease, but this too has not improved prognosis.
Commonly, the disease is advanced at initial presentation with regional nodal spread in up to 20 % of patients and systemic metastases in up to 40 % [87, 88, 90–93, 96]. Five-year overall survival is estimated at 20–22 % with a disease-free survival of 16–17 % [87–90, 92, 93, 100, 102]. Recurrence is often systemic and fatal. A predictor of poor outcome is thought to be the presence of perineural invasion in the primary tumor; however, unlike cutaneous melanoma, lymph node status has not shown an impact on survival [95, 100, 103].
Due to the uniformly poor prognosis of ARM, efforts combining surgical resection with effective systemic therapy will be required for the successful management of this disease. Recently survival outcomes have improved in the treatment of cutaneous metastatic melanoma through advances in both immunotherapy and targeted therapy. Ipilimumab, a monoclonal antibody targeting cytotoxic T-lymphocyte antigen 4 (CTLA-4), was the first agent to improve overall survival in patients with metastatic melanoma in a phase III, randomized, control trial [104, 105]. Additionally, vemurafenib, an inhibitor of mutant BRAF, has increased both progression-free and overall survival in a phase III trial in patients with melanoma containing the V600E BRAF mutation [106]. Lastly, various inhibitors of KIT, such as imatinib mesylate, sunitinib, nilotinib and dasatinib, have also demonstrated survival benefit in a subset of patients with metastatic melanoma harboring mutant KIT [107]. BRAF and KIT mutation rates vary in mucosal melanomas according to anatomic site [108, 109]. Thus, it can be hypothesized that those ARM which are more immunogenic may represent better targets for drugs such as ipilimumab which exert its effect through immune system modulation, while those which possess KIT or BRAF mutations may demonstrate a therapeutic response to targeted inhibitors, such as imatinib and vemurafenib. While no current study has specifically examined ARM treatment with these new drugs, new trials utilizing these agents, including ipilimumab, BRAF- and KIT-inhibitors, for the treatment of all mucosal melanomas, are underway.
Neuroendocrine Carcinoma (NEC) of the Rectum
Neuroendocrine tumors (NETs) of the rectum are rare, comprising 0.2–0.4 % of colorectal malignancies [110–112]. Neuroendocrine neoplasms have been described in multiple organ systems, including respiratory, genitourinary and endocrine organs in addition to the gastrointestinal tract. The most common location of NETs in the large bowel is the rectum, followed by the cecum and sigmoid [111–115]. These lesions possess an endocrine function in that they are able to synthesize and secrete a multiple amines and hormones, including several neurotransmitters. Through advancements in both immunohistochemistry and microscopy GI neuroendocrine tumors were further categorized by degree differentiation [110, 114, 116, 117] and it is now known that many different rectal neoplasms (including carcinoids, certain anaplastic tumors, and small cell tumors) display neuroendocrine features [110]. Thus, GI neuroendocrine tumors incorporates a spectrum of well to poorly differentiated tumors of various sizes. The term “carcinoid” has been used to describe a subgroup of smaller, indolent, well-differentiated GI NETs while neuroendocrine “carcinoma” represented the poorly differentiated, more aggressive lesions in this group. This naming taxonomy still lacked clinical significance, however, because even small, low-grade carcinoids may metastasize (see section on “Carcinoid tumors of the rectum”).
Recently, there has been improvement in both classification and prognostic value of these tumors, beginning with development of the 2010 WHO guidelines which grades NETs based upon degree of differentiation as determined by immunohistochemical features of mitoses and Ki-67 index (see Table 22.2) [118, 119]. Carcinoids are considered to be low or intermediate grade NETs of the colon and rectum (Grade 1 or 2), or well-differentiated lesions. Grade 3 or poorly differentiated lesions are considered neuroendocrine carcinomas of small cell type, and less frequently large cell carcinoma. This has since further been elaborated upon by the American Joint Cancer Committee (AJCC) and the European Neuroendocrine Tumor Society (ENETs) who have developed a Tumor, Nodal, Metastasis staging system (TNM) in accordance with the WHO classification [16, 120, 121]. Validation studies confirmed that TNM staging systems accurately stratify colorectal NETs in a prognostically significant way [122, 123]. Additionally, the WHO grading guidelines, based on the Ki67 proliferative index, demonstrated statistically significant different survival outcomes between all grades on multivariate analysis further confirming the value of this classification system [124]. Thus, this separation of colorectal NETs into prognostically relevant subgroups by the TNM staging and WHO grading systems may aid in more streamlined, standardized treatment of these lesions in the future.
Table 22.2
The WHO 2010 Gastrointestinal Neuroendocrine Tumor (NET) grading classification
Morphology | Mitotic count | Ki67 index (%) | |
|---|---|---|---|
Grade 1 | Low grade | <2/10 HPF | ≤2 |
Grade 2 | Intermediate grade | 2–20/10 HPF | 3–20 |
Grade 3 | High grade | >20/10 HPF | >20 |
High-grade tumors or neuroendocrine carcinomas (NECs) contain abundant necrosis, either confluent or punctate within nests of tumor cells where as low-grade neuroendocrine neoplasms (e.g., carcinoid) generally possess some degree of typical native organ architectural patterns [125]. Up to half of colorectal NETs may contain non-neuroendocrine elements such as squamous or adenocarcinoma components [112, 114, 115, 117]. The pathogenesis of these tumors is not clear. One hypothesis is that pluripotent stem cells within the colonic epithelium exist and undergo malignant transformation leading to NE differentiation. Conversely, another hypothesis is that some adenocarcinomas, during malignant transformation, may develop NE characteristics. Additionally, there are genetic similarities between colorectal adenocarcinomas and highly aggressive NECs which are not shared by the more benign, well-differentiated carcinoid tumors. This suggests that there may not be a common origin between these subtypes of neuroendocrine tumors despite ultrastructural and immunohistochemical similarities. Loss of heterozygosity for the APC (adenomatous polyposis coli), DCC (deleted in colorectal carcinoma), or p53 genes, which are seen in adenocarcinomas are common for NECs, but are not associated with well-differentiated carcinoid lesions [125]. The exact reasons behind this remain unclear.
Clinical Presentation and Diagnosis
Most patients diagnosed with NETs of rectum are in their 50s–60s [5, 9–12, 16]. There is a slight male predominance and higher rates of rectal NETs among black and Asian populations [3, 5]. The incidence of colonic and rectal NETs may be increasing, perhaps due to the more frequent use of endoscopy for diagnosis and screening purposes [3, 5–7]. Most patients with rectal NETs are asymptomatic and are diagnosed incidentally in patients undergoing screening or endoscopic testing for unrelated reasons [10, 17].
Abdominal imaging (CT or MRI) combined with lower endoscopy provides significant information to aid in the staging and diagnosis of colorectal NETs. Endoscopic Ultrasound (EUS) has also played a key role in staging and treatment of rectal NETs; by providing data on size, depth of invasion, and lymph node involvement EUS helps determines the feasibility of conservative (endoscopic or transanal excision) management or the necessity of radical surgery (LAR or APR) [19, 25]. Electron microscopy and immunohistochemistry, are often required however for definitive diagnosis. Immunohistochemistry provides information on NE differentiation and bioactive amines. Rectal NE carcinomas, like rectal carcinoids, seem relatively incapable of producing carcinoid syndrome (even with liver metastases). Lastly, when small cell NEC of the rectum is diagnosed on rectal biopsy, which is histologically identical to small cell carcinoma of the lung, a search to rule out a pulmonary primary tumor must be performed.
Treatment and Prognosis
High-grade colorectal NETs, unlike low or intermediate grade carcinoids, are extremely aggressive tumors with poor prognosis. This tumor is associated with a 58 % 6-month survival rate, and a 6 % 5-year survival rate in some series [16, 110]. All rectal NETs, including well-differentiated tumors, have an overall 5-year survival of 88.3 %, with localized disease having a rate of 90.8 %, regional disease at 48.9 % and those with distant metastases 32.2 % [3]. This finding reflects that most of rectal carcinoid tumors (82 %) are localized at diagnosis, with a median size of only 0.6 cm [123]. Colon NECs proximal to the rectum are more aggressive on average, with a 5-year survival of only 62 % across all stages [3]. These tumors have a high propensity for nodal and distant metastasis. As high as 65–80 % of patients will have nodal or distant metastasis at presentation [110, 126, 127]. Tumor grade, tumor size, depth of invasion, lymphovascular invasion, elevated mitotic rate and lymph node involvement significantly predict malignant behavior in localized rectal NETs [128]. According to one analysis of the literature, metastases were observed in 2 % of patients with rectal NETs measuring less than 1.0 cm, 10–15 % of tumors measuring 1.0–2.0 cm, and 60–80 % in patients with tumors measuring greater than 2.0 cm [129]. Patients appear to have a marginally better prognosis if they present without metastatic disease, have an adenocarcinoma component within their tumor, or respond to chemotherapy. Surgery, particularly in the presence of metastatic disease, may not offer a survival benefit for the majority of patients [127].
There is no standardized management of rectal NETs; as with carcinoids, treatment has been guided by tumor size. Because of their low risk of metastatic spread, localized disease or tumors that are small (<1 cm) and confined to the mucosa or submucosa (T1) can be managed with endoscopic resection or transanal excision. In lesions of 1–1.9 cm, transanal endoscopic microsurgery (TEMs) should be considered, which allows for deeper, full-thickness excision [130]. Larger tumors and those with adverse features should undergo APR or LAR. Palliative resection in advanced disease may be offered particularly where debulking may improve symptoms or relieve obstruction.
There are no good treatment outcomes for patients with metastatic colorectal NETs [16]. Metastatic hindgut NETs are incurable and optimal management requires a multidisciplinary approach with chemotherapy [131]. Palliative chemotherapy with or without radiation therapy has been used for adjuvant therapy and for treatment of metastatic disease without a clear survival benefit. For those few hindgut patients with functional tumors, somatostatin analogs are effective in the management of carcinoid syndrome and may delay disease progression. Liver-directed therapy and surgical debulking can improve the quality of life for some patients.
Due to the high rates of recurrence in colonic and rectal NETs, they require surveillance for recurrence even after successful complete resection at surgery except where the risk of recurrence is very low i.e. pT1a lesions <1 cm. There is little common consensus as to the best surveillance modality, interval period or length of surveillance but the European Neuroendocrine Tumour Society (ENETS) has published guidelines based upon tumor size [120]. Lesions <1 cm that are well differentiated (G1) with no invasion of the muscularis or lymphovascular invasion require no follow-up if resection is complete. Lesions that are between 1 and 2 cm or are higher grade should undergo annual follow-up with endoscopy, EUS and MRI. Lesions that are >2 cm: one endoscopy, CT or MRI scan, and serum marker within the first year. For high grade (G3) tumors: surveillance endoscopy, CT scan and serum marker every 4–6 months in the first year, and thereafter at least annually. ENETS recommends follow-up for at least 10 years [120], while NANETS guidelines recommend surveillance for up to 7 years [16].
Vascular Lesions
Vascular lesions occur throughout the GI tract and frequently present in a delayed fashion after repeated episodes of bleeding and unwarranted procedures. The nomenclature of vascular lesions has been tainted by confusion and misnomers, but two main categories of vascular lesions have emerged: hemangiomas and vascular malformations [132]. Hemangiomas are usually absent at birth but appear at 6–8 weeks of life. Their course is marked by rapid proliferation followed by spontaneous involution. They have a high endothelial cell turnover. Vascular malformations, however, are usually present at birth, have normal endothelial cell turnover and grow in proportion with the person [133]. Vascular malformations are further classified according to their dominant abnormality into arteriovenous, venous, lymphatic, lymphatic-venous, and capillary malformations. The term hemangioma is often erroneously used to describe vascular malformations in the GI tract. The commonly used term “cavernous hemangioma” will here be replaced by the more correct term “cavernous malformation” or “diffuse cavernous malformation.” Hemangiomas do occur in the GI tract but far more infrequently than vascular malformations. Intestinal vascular malformations are classified into capillary, cavernous (localized or diffuse infiltrating), mixed, and hemangiomatosis. Of rectosigmoid malformations, 80 % are of the cavernous type. The localized type of cavernous malformations is frequently polypoid and symptomatic while the diffuse type can be multiple and has been reported up to 30 cm in length. Some types can be circumferential and invade surrounding structures.
Diffuse Cavernous Malformation
Diffuse cavernous malformation (previously diffuse cavernous hemangioma) of the rectum is a rare condition albeit an important differential diagnosis in rectal bleeding. Approximately 130 cases have been described in the literature [133]. The first case was described in 1839 by Buie and Nesselrod in their paper “Erectile tumor of the anus.” Diffuse cavernous malformations comprise approximately 20 % of intestinal angiomas [134]. They can be found anywhere in the GI tract but occur most commonly in the rectosigmoid area, or in 50–70 % of cases [135].
Pathology
The malformation can be limited to a small area or be diffuse and infiltrate adjacent structures such as the bladder or uterus. The diffuse cavernous malformations are of variable sizes, sometimes up to 20–30 cm in length and are occasionally found in multiple locations [134]. These lesions do not have malignant potential and might be better classified as hamartomatous rather than neoplastic lesions. Malignant degeneration is exceedingly rare. The diffuse infiltrating cavernous hemangioma may replace the bowel wall from serosa to mucosa. Histologically, these malformations are composed of multiple dilated tortuous vessels within a stroma containing abundant smooth muscle and fibrous connective tissue [136]. Diffuse cavernous malformations are associated with Klippel-Trénauny syndrome [137] and Kasabach-Meritt syndrome [138].
Clinical Presentation and Diagnosis
The disease is characterized by recurrent, moderate-to-massive, and sometimes fatal, gastrointestinal bleeding. Blood transfusions are frequently required. The first episode usually occurs in childhood or infancy [139]. The mean age at diagnosis was 6.5 years in Londono-Schimmer’s series of 15 patients [140]. A delay of several years from initial presentation to diagnosis is not uncommon and up to 80 % of patients have undergone at least one unwarranted surgical intervention in an attempt to correct their condition, most often a hemorrhoidectomy [139, 141, 142]. The bleeding is usually painless and often intensifies with each successive episode [135]. Approximately 17–25 % of patients present with obstructive symptoms from voluminous growth into the bowel lumen, intussusception, or occasionally, volvulus. Rarely, a rectal cavernous malformation may cause tenesmus, urgency and incomplete evacuation [134]. Although the diagnosis is frequently delayed, it can be suspected from a family history or personal history of mucocutaneous lesions, anemia, frank bleeding or signs of obstruction. The lesion may be palpable on rectal exam and other vascular lesions of the mucous membranes or skin may be seen.
This vascular neoplasm is usually diagnosed by one of several different modalities. Endoscopy is diagnostic and frequently shows nodular masses which are soft and compressible and deep blue, purple or dull red in color [143]. Biopsies are generally contraindicated due to the risk of severe bleeding. Phleboliths are normally not seen on abdominal x-ray in younger individuals but are seen in 50 % of plain x-rays in patients with diffuse cavernous malformations [144]. Phleboliths in unusual areas in the pelvis in a person with history of rectal bleeding and constipation should prompt suspicion of diffuse cavernous malformation. Barium enema can show irregularity, nodularity and obstruction. CT is an excellent diagnostic modality that reveals both the extent of malformation and possible invasion into adjacent structures. CT findings include marked transmural thickening of the bowel and mesentery, heterogeneous enhancement, bowel narrowing, nodular indentations of the rectosigmoid wall, and anterior displacement of the rectum [145]. Angiogram shows the lesion well and can localize active bleeding but is invasive and has been replaced by more modern imaging modalities such as CT or MRI. MRI has excellent soft tissue discrimination, has the ability to show blood flow without the use of intravenous contrast, and does not use ionizing radiation [145]. Phleboliths, however, are poorly detected on MRI [145]. Ultrasound is also a useful imaging modality but is user dependent.
Treatment and Prognosis
The treatment of diffuse cavernous malformations is surgical. Fatal bleeding in untreated patients was reported as high as 45 % in a small series [145]. Treatments such as embolization, radiotherapy and sclerotherapy have thus far been unsuccessful. Sphincter-saving resection with coloanal anastomosis with radical removal of the abnormal tissue is the procedure of choice in these usually young patients with benign disease [146, 147]. Abdominoperineal resection with a permanent end colostomy was used with relatively low morbidity and mortality in the early patients [148]. This soon fell out of favor for sphincter-preserving resections, now the procedures of choice. Colonic pull-through procedures have been performed with some success, like the Soave procedure [140, 149], and a modified Soave procedure [150]. Small polypoid cavernous malformations usually are limited to the submucosa and have been safely removed with endoscopic cautery snare polypectomy [151]. However, the safety of this intervention is unclear and massive hemorrhage could ensue. The following criteria for endoscopic polypectomy have been proposed: size ≤2.5 cm, pedunculated or sub-pedunculated polyp, and depth of lesion limited to the submucosa [143].
Lymphangioma
Rectal lymphangiomas are exceedingly rare with only a few reported cases to date [152]. These lesions consist of abnormal dilatation and mass-like proliferation of lymphatic channels. They have been incidentally detected during colonoscopy, but when symptomatic, present with pelvic pain and rectal bleeding [153, 154]. On colonoscopy, they frequently appear pedunculated or as cystic submucosal nodules with a smooth, translucent surface that is easily compressible. Lymphangiomas less than 20 mm in size can be removed by snare polypectomy but larger, sessile, and infiltrating lesions may require surgical resection [155].
Hemangiopericytoma
Hemangiopericytoma is a rare tumor first described in 1942 by Stout and Murray [156]. It is comprised of profuse proliferation of capillaries surrounded by sheets of rounded, sometimes elongated pericytes [157]. They can become quite large and cause compressive symptoms, obstruction, intussusception and gastrointestinal bleeding [158]. These tumors are variable in their behavior and occur in all age groups without a gender predilection. While some tumors remain localized for decades, some are malignant, with approximately a third developing aggressive invasion or metastases, more commonly in older patients [159, 160].
Metastases can be detected years after removal of the original tumor. Out of 106 cases reported in 1976, 26 were in the retroperitoneum and pelvis [161]. Seven cases of colorectal hemangiopericytoma were reported in 1959 [162]. Their size ranged from 3.5 to 26 cm in greatest dimension. Local recurrence and distal metastases were common.
Squamous Cell and Adenosquamous Carcinoma of the Rectum
Squamous Cell Carcinoma of the Rectum
Squamous cell carcinoma of the colon and rectum is extremely rare with approximately 70 cases reported in the literature [166], comprising approximately 0.1 % of all colorectal neoplasms [167]. The rectum harbors nearly half of these tumors. Williams et al., in 1979 set forth criteria for the diagnosis of primary colorectal squamous cell carcinoma: (1) The lesion may not be a secondary metastasis from another primary lesion. (2) There should be no squamous-lined fistula track, where squamous carcinoma is known to originate. (3) A rectal squamous cell carcinoma should not be an extension from an anal squamous cell primary [168].
The rectum normally does not contain squamous cells, but several theories have been proposed to explain the development of squamous cell carcinoma in this location: (1) Proliferation of pluripotent stem cells, which exist in the mucosal crypts following mucosal injury. (2) Squamous metaplasia resulting form chronic irritation [169], although squamous metaplasia is rarely seen; similarly squamous cell carcinoma is rarely seen. (3) Persistent embryonal nests of committed or uncommitted ectodermal cells remaining in an ectopic site after embryogenesis. This could explain the lower rectal squamous carcinomas, but is unlikely to explain colonic tumors. (4) Squamous differentiation arising in an adenoma. Rare adenomas have been found to have squamous differentiation, which could indicate the parallel adenoma-carcinoma sequence of squamous cells [168, 170]. Squamous carcinomas have clinicopathological features in common with adenocarcinomas; the age and sex distribution of affected patients are similar for adenocarcinoma and squamous cell carcinoma; the anatomic distribution of adenomatous and squamous tumors is similar within the large bowel, although the number of squamous cell tumors is too low to allow for a meaningful statistical correlation. No association has been found between HPV subtypes 6, 11, 16 and 18 and squamous or adenosquamous carcinoma [171]. Immunohistochemical staining of keratin suggests a pluripotent endodermal stem cell origin for both squamous call carcinoma and adenocarcinoma of the rectum [172].
Clinical Presentation and Diagnosis
Squamous cell carcinoma of the rectum usually presents in the fifth decade of life (range 33–93) and more often in women than in men [172]. Symptoms at presentation are similar as with adenocarcinoma, such as abdominal pain, hematochezia, diarrhea, constipation, anorexia and weight loss. Lafreniere et al. reported positive lymph nodes in 57 % of patients at diagnosis [173]. Distant metastases at diagnosis were found in 21 % of patients to either lungs, liver, vertebrae, omentum mesentery, peritoneum or adrenal glands [173]. Concomitant conditions such as ulcerative colitis, colonic duplication, schistosomiasis, amoebiasis, ovarian cancer, endometrial cancer, ovarian teratoma, prostate cancer, and a chronic colocutaneous fistula have all been reported but a causative relationship is uncertain [167, 170]. A tenth of patients were found to have antecedent, synchronous or metachronous adenocarcinoma [167]. Although no definitive conclusion can be drawn from this due the rarity of the disease, it suggests that clinicians should maintain a heightened awareness for the risk of other neoplasms.
Diagnosis is obtained with physical exam and endoscopic examination with biopsies. Tumor and nodal staging can be further achieved by endorectal ultrasound. The presence of distant metastatic disease is evaluated by X-ray or CT of the chest, and abdominopelvic CT.
Treatment and Prognosis
The optimal treatment for these rare tumors is not clearly defined but has been primarily surgical in the form of segmental resection or abdominoperinal resection. Primary palliative chemotherapy was reported in one patient with some response [174]. The pendulum seems to be swinging towards non-surgical management with the first-line treatment being combination chemoradiation therapy as described by Nigro and is now the standard of care for squamous cell anal cancer [175, 176]. Case reports and case series suggest that rectal squamous cell carcinoma responds well to initial chemoradiation therapy, with surgery reserved for salvage treatment of non-responders, partial responders or for recurrence [173, 177–179]. The need for subsequent surgery is unclear, but a sphincter-preserving surgery should be feasible in most cases [172].
Possibly due to the rarity of colorectal squamous cell carcinoma, initial data suggested a poor prognosis with 30 % 5-year survival [180]. When examining a larger number of cases, the prognosis seems to be similar stage-for-stage to node-negative colorectal adenocarcinoma (stages I and II). The prognosis is worse, however, when nodal disease occurs in SCC than for adenocarcinoma of a similar stage [166]. Features that predict a poor prognosis include right-sided colon lesions, ulcerated or annular cancers, node-positive disease, grade 3 or 4 cancer, and stage IV disease [166].
Adenosquamous Carcinoma of the Rectum
Adenosquamous carcinoma is an extremely rare malignancy with an incidence of 0.025–0.1 % of all colorectal cancers [181]. As its name implies, it has both glandular and squamous histologic components, both of which are malignant and can metastasize. The mean age at diagnosis of adenosquamous cell carcinoma is in the sixth and seventh decades with an equal gender distribution [167, 182]. Although Cagir et al. found the rectum and distal colon to be affected most often [181], others have found adenosquamous carcinoma more frequently in the right colon [166, 182].
The cause of adenosquamous lesions is unknown, but the theories for histogenesis mirror the ones for squamous colorectal cancer. There may be an association with ulcerative colitis [183], polyposis, schistosomiasis, ovarian adenocarcinoma and endometrial adenocarcinoma [184]. The squamous component seems to metastasize more frequently and is more aggressive than the glandular component [184].
Adenosquamous carcinoma seems to be more aggressive than adenocarcinoma and have a worse prognosis stage for stage. Cagir et al. also found that 85 % of their patients presented with regional or metastatic disease [181].
The primary treatment modality for these tumors is surgery. Adjuvant chemotherapy is frequently used but the benefit is unknown in this uncommon disease [185]. The Nigro regime has also been used as an adjunct after surgery [181].
In a recent population based study using the California SEER database, Masoomi et al. identified 99 cases of adenosquamous carcinoma [182]. They found that adenosquamous tumors present with more advanced disease and more poorly differentiated tumors. The 5-year survival was worse compared to adenocarcinoma with an increased overall mortality. They concluded hat adenosquamous tumors should be considered a poor prognostic factor in patients with colorectal cancer. The overall 5-year survival is 30 % or less [182] and the mean survival is 12 months [181].
Sarcomas of the Rectum
Leiomyosarcoma of the Rectum
Sarcomatous tumors of the colon and rectum are rare and include tumors such as fibrosarcoma, angiosarcoma, leiomyosarcoma and GIST. Before the discovery that most leiomyosarcomas are in fact gastrointestinal stomal tumors, 95 % of colorectal sarcomas were attributed to leiomyosarcomas. Their earlier described incidence of 0.07–0.1 % of all rectal malignant tumors is in fact much lower [186]. They are most common in the lower third of the rectum [186, 187]. They remain difficult to diagnose and are very aggressive with a poor prognosis.
While GISTs arise from interstitial cells of Cajal and express KIT, leiomyosarcomas have a distinct lineage from smooth muscle cells and do not express KIT. Our previously held notions on leiomyosarcoma need to be redrafted with newer studies using modern diagnostic criteria, based on findings from true leiomyosarcomas rather than findings from GISTs.
Pathology
Leiomyoma and leiomyosarcoma are found throughout the GI tract with 7 % of them in the rectum [186]. Leiomyosarcomas are usually larger than leiomyomas; they are soft to rubbery firm, and frequently very vascular [188]. They arise from smooth muscle in the muscularis mucosa, round or longitudinal muscle of the bowel wall, or from the vascular smooth muscle [188]. Malignant degeneration of benign leiomyoma has been described, although the exact pathogenesis is not known [188, 189].
Histologically, leiomyosarcomas appear as well-differentiated smooth muscle cells, composed of elongated cells growing in fascicles with at least focal pleomorphism and high mitotic activity [190]. Microscopic differentiation between leiomyoma and leiomyosarcoma can be very difficult [188]. Immunohistochemically, they are positive for actin and desmin and negative for KIT and CD34, differentiating them from GIST tumors [190]. Based on the number of mitoses per 10 consecutive high-power fields, the tumor is classified into high-grade (10≥/10 HPFs) or low-grade tumor (<10 mitoses/10 HPFs) [191].
Spread is local, with direct invasion, or blood-borne, most often to lungs and liver, but also to peritoneum, brain and spine. Metastases to lung and liver is the most common cause of death [186]. Although it is generally said that leimyosarcoma does not spread to lymph nodes [191], there are case reports of lymph node involvement, mainly with very poorly differentiated tumors [188].
Clinical Presentation and Diagnosis
The tumor occurs more frequently in males, while benign leiomyomas tend to occur more frequently in females [188]. The tumor is most often diagnosed in the fifth and sixth decades. Presenting symptoms include bleeding, constipation, rectal pain, sense of fullness and diarrhea [186]. Most tumors appear as submucosal masses that mainly grow into the lumen but also can grow away from it [186]. The tumor may protrude into the lumen or partially occupy the circumference of the rectum. Approximately 50 % of the tumors are ulcerated [188]. Khalifa et al. carefully reviewed 136 cases of rectal leiomyosarcoma [186]. The majority of tumors were in the lower rectum, and 89 % of them were palpable by rectal exam. The tumor size ranged from 1 cm to 15 × 10 × 20 cm but tumors up to 30 cm have been identified [192]. Diagnosis is made by endoscopy and biopsy, although biopsies can be difficult to obtain [188].
Treatment and Prognosis
The mainstay of treatment is surgical. Any clear recommendation regarding the choice of treatment is difficult for this rare malignancy, but a few trends have emerged from several case reports and retrospective series [186, 187, 193]. It is important to recognize the selection bias when comparing the different surgical approaches as patients are selected for surgery based on size and state of their primary tumor. Local excision has been followed by high local recurrence rates up to 60–67 %, necessitating a more aggressive surgical approach [186, 194] The local recurrence rate after abdominoperineal resection is much lower, 20–24 %, but this has not translated into a difference in long-term survival when comparing the two general surgical modes [186, 187, 192, 194]. The disease-free survival at 5 years was 32 % after wide local excision and 52 % after radical resection but overall survival was identical [187].
Randleman Jr et al. concluded that anorectal lesions smaller than 2.5 cm in diameter and confined to the bowel wall could be treated with wide local excision and patients with larger, non-confined lesions might do better with a more radical resection [193].
Adjuvant radiotherapy was used early on but was soon believed to be of little benefit [188, 195]. No benefit of either radiation treatment or chemotherapy has been shown [193].
The overall prognosis for patients with rectal leiomyosarcoma is poor with a 5-year survival of 40 % [196] and median survival of 33 months [192]. A young age at diagnosis (under 50 years) and a high histologic tumor grade have been found to be poor prognostic factors, stressing the need to identify adjuvant treatments for these patients [187, 192, 194]. Recurrence has been noted 15–17 years after treatment, underlining the need for long-term follow-up [192].
Rectal Gastrointestinal Stromal Tumor
Gastrointestinal stromal tumors (GISTs) have been recognized as the most common mesenchymal tumors of the GI tract. Prior to the advent of immunohistochemical diagnostics, most GI stromal tumors were identified as leiomyosarcoma. GISTs occur in 1.1–2 persons per 100,000 [197]. The rectum is the third most common site for GIST, accounting for 4–5 % of all GIST tumors [190, 198]. The tumor occurs more commonly in males and usually between the fifth and seventh decades of life [190].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


