Kidney transplantation is universally accepted as the therapy of choice for children with end-stage renal disease (ESRD). About two thirds of pediatric patients with ESRD ultimately receive a kidney transplant. Successful transplantation in children and adolescents not only ameliorates uremic symptoms but also allows for significant improvement of delayed skeletal growth, sexual maturation, cognitive performance, and psychosocial functioning. The child with a well-functioning kidney transplant can enjoy a quality of life that cannot be achieved by any form of dialysis therapy.
Current success in pediatric renal transplantation is attributed to improvements in transplantation technology, immunosuppressive therapy, and the provision of age-appropriate clinical care. Transplantation continues to result in better survival than dialysis for pediatric patients of all ages. Five-year survival rates in transplant patients are close to 95%, whereas in dialyzed patients, the survival rates are about 80%. Nevertheless, success in pediatric kidney transplantation remains a challenging undertaking. Children and adolescents are constantly growing, developing, and changing. Each developmental stage produces a series of medical, biologic, and psychological challenges that must be appropriately addressed if truly successful graft outcome and rehabilitation are to be realized.
Much of the statistical data reviewed in this chapter comes from databases that have provided an invaluable resource for the advancement of pediatric transplantation. These databases have enabled the evaluation and extrapolation of data from multiple pediatric renal transplant programs that tend to be small when compared with their adult counterparts. Major databases referred to are the North America Pediatric Renal Transplant Cooperative Study (NAPRTCS), the Scientific Registry of Transplant Recipients (SRTR), and the United States Renal Data System (USRDS) annual report (see websites).
EPIDEMIOLOGY OF END-STAGE RENAL DISEASE IN CHILDREN
Incidence
The incidence and prevalence of treated pediatric ESRD have been increasing since 1989. As of 2005, the incidence rate of new cases of ESRD in children up to 19 years of age was 15 per 1 million U.S. children per year. The point prevalence of ESRD in this population is 80 per 1 million U.S. children. The incidence of ESRD increases with age, with the highest incidence observed in children between 15 and 19 years of age (28 per million). Adolescents represent approximately 50% of treated pediatric ESRD patients.
There is a wide variation by ethnic group in the incidence rates of treated ESRD. African American children have the highest incidence rate of 27 per 1 million, as compared with 12 per 1 million white children, 15 per 1 million Asian and Pacific Islander children, and 17 per 1 million Native American children. The incidence is higher in African Americans across all age groups but is most prominent in the 15- to 19-year-old age group (60 per 1 million African Americans compared with 20 per 1 million whites). During the past 20 years, incident rates for white pediatric patients have remained constant. For African American patients and other nonwhite ethnicities, however, the rates of ESRD have more than doubled. The incidence of glomerulonephritis as a cause of ESRD is up to 3 times higher in African American pediatric patients than in white pediatric patients; there is no racial predilection for other causes of pediatric renal disease. Patients with focal segmental glomerulosclerosis (FSGS) make up almost 23% of all pediatric African American dialysis patients and more than 30% of adolescent African American dialysis patients. Boys have higher incidence of treated ESRD than girls in all age groups.
Etiology
Congenital, hereditary, and cystic diseases account for about 50% and glomerular diseases for 20% of cases of pediatric ESRD (Table 16.1). Incidence rates for patients with glomerular diseases and patients with congenital, hereditary, and cystic diseases continue an upward trend.
The most common primary diagnoses remain aplastic, hypoplastic, or dysplastic kidney and obstructive uropathy, each present in about 16% of patients. FSGS is the third most common (12%) and continues to be the most prevalent acquired renal disease. In contrast to adults, ESRD caused by diabetes mellitus or hypertension is rare in children.
Access to Transplantation
Between 1987 and 2007, more than 9,500 children received 10,400 transplants in the United States. At the time of transplantation, about two thirds of pediatric recipients of kidney transplants are older than 12 years, 17% are 6 to 12 years of age, 17% are between the ages of 2 and 5 years, and less than 1% are younger than 1 year of age. About 60% are male, 61% are white, 17% are African American, and 16% are Hispanic.
Pediatric transplantations constitute 4% to 7% of all kidney transplantations in the United States. The number of pediatric kidney transplantations has remained relatively constant during the past decade at about 800 each year. During the same period, the number of adult kidney transplantations has increased by nearly 50% (see Chapter 1, Fig. 1.1). Historically, the number of living donor transplants consistently exceeded the number of deceased donor transplants in pediatric ESRD. As of 2005, the number of pediatric kidney deceased donor transplants exceeded living donor transplants, following the trend found in adults. The number of deceased donor transplants increased from 278 in 2000 to 468 in 2005. This trend is likely a result of changes made to the United Network for Organ Sharing donor allocation policy, whereby children receive priority for deceased donor kidneys from donors younger than 35 years (see Chapter 4).
Children continue to represent only a small percentage of the national waiting list for deceased donors. During the past decade, their number has remained constant in the range of 500 to 650, whereas the adult list has more than doubled in number. Additionally, median waiting times have decreased in pediatric patients. In 2005, the median waiting time for all pediatric groups was less than 300 days. This compares favorably with the latest data on median waiting time in adults, which during the past 10 years was never less than 920 days in any age group. This difference reflects kidney allocation rules that are specifically designed to favor children because of their unique needs for growth and development.
TABLE 16.1 Incidence of End-Stage Renal Disease in Pediatric Transplant Patients According to Primary Disease, 2007*
Primary Renal Disease
Incidence (%)
Cystic, hereditary, and congenital disease
47.4
Renal hypoplasia, dysplasia
16.0
Congenital obstructive uropathy
15.6
Polycystic disease
3.0
Medullary cystic disease (nephronophthisis)
2.8
Prune belly syndrome
2.6
Congenital nephrotic syndrome
2.6
Alport syndrome, other familial disease
2.2
Cystinosis
2.1
Oxalosis
0.5
Glomerulonephritis (GN)
21.3
Focal segmental glomerulosclerosis
11.7
Chronic GN
3.4
Membranoproliferative GN type I
1.8
Idiopathic crescentic GN
1.8
IgA nephropathy
1.3
Membranoproliferative GN type II
0.9
Membranous nephropathy
0.4
Interstitial nephritis, pyelonephritis
7.1
Chronic pyelonephritis, reflux nephropathy
5.3
Interstitial nephritis
1.8
Secondary GN, vasculitis
6.2
Hemolytic uremic syndrome
2.7
Systemic lupus erythematosus
1.5
Henoch-Schbönlein purpura
1.2
Wegener granulomatosis
0.5
Other systemic immunologic disease
0.3
Hypertension
1.3
Miscellaneous conditions
1.3
Neoplasms
1.0
Sickle cell nephropathy
0.2
Diabetes mellitus
0.1
Other
9.4
Uncertain etiology
6.0
*The study included 9506 patients younger than 21 years.
Modified from the NAPRTCS 2007 annual report available at www.emmes.com/study/ped.
Timing of Transplantation
Renal transplantation should be considered when renal replacement therapy is indicated. In children, dialysis may be required before transplantation to optimize nutritional and metabolic conditions, to achieve an appropriate size in small children, or to keep a patient stable until a suitable donor is available. Many centers prefer that a recipient weigh at least 8 to 10 kg, both to minimize the risk for vascular thrombosis and to accommodate an adult-sized kidney. In infants with ESRD, a target weight of 10 kg may not be achieved until 12 to 24 months of age. At some centers, transplantation has been successful in children who weighed less than 10 kg or who were younger than 6 months.
Preemptive transplantation (i.e., transplantation without prior dialysis) accounts for 25% of all pediatric renal transplantations. The major reason cited by patients and families for the decision to undertake preemptive transplantation is the desire to avoid dialysis. Candidates for preemptive transplantation should have careful psychological assessment before transplantation because there may be a greater tendency for noncompliance in children who have not experienced dialysis. Nevertheless, there appears to be no impairment in graft outcome in pediatric recipients who have undergone preemptive transplantation when compared with those who have undergone dialysis before transplantation, and some data suggest a small improvement in allograft outcome. The reasons for the improved graft survival are unknown. Because of the waiting time for deceased donors, most preemptive kidney transplants are from living donors.
Patient and Graft Survival
Both patient and graft survival rates have improved steadily since systematic recording began in 1987. Patient survival after transplantation remains superior to that achieved by dialysis for all pediatric age groups. The overall 1- and 5-year patient survival rates are now 98% and 94%, respectively, for all primary transplants, and are marginally better for recipients of living donors than for deceased donors. The 1- and 5-year patient survival rates for living donor recipients are 98% and 96%, whereas those for deceased donor recipients are 97% and 93%. Although patients younger than 2 years of age have lower survival rates, the situation has significantly improved within the past decade. Infection accounts for 29% of deaths. Other causes include cardiopulmonary disease (15%), malignancy (11%), and dialysis-related complications (3%). About 47% of patients who die do so with a functioning graft.
Graft survival rates for pediatric transplants are somewhat better than for adult transplants. One- and 5-year graft survivals are 95% and 85%, respectively, for living donor recipients and 93% and 77%, respectively, for deceased donor recipients. Of the more than 10,000 pediatric kidney transplantations performed since 1987, about 26% have failed. Chronic rejection accounts for 40% of graft failures, with acute rejection accounting for 9%. Other causes include vascular thrombosis (8%), recurrence of original disease (8%), and patient noncompliance (6%). Chronic rejection remains the most common and ever-increasing cause of allograft failure. Although some causes of graft failure, such as recurrence of the original disease, have remained constant during the past 10 years, loss from acute rejection and graft thrombosis has decreased. Technical issues remain a challenge and are a more common cause of graft loss in children than in adults.
PROGNOSTIC FACTORS INFLUENCING GRAFT SURVIVAL
The following factors are important determinants of the improving graft survival reported in pediatric patients. Long-term renal function is a particularly important consideration in pediatric renal transplantation because of its impact on post-transplantation skeletal growth.
Donor Source
Short- and long-term graft and patient survival rates are better in recipients of living donor transplants in all pediatric age groups. Younger transplant recipients benefit the most from living donor transplantation and enjoy a 20% to 30% better graft survival rate 5 years after transplantation. Shorter cold ischemia time, better human leukocyte antigen (HLA) matches, lower acute rejection rates, and better preoperative preparation help to account for the improved outcome. During the past decade, however, marked improvement has been made in deceased donor patient and graft survival. These results may be related to temporal trends in immunosuppressive drugs, decreased transfusion requirements, and decreased use of young deceased donors.
Recipient Age
The trend for younger children, especially those younger than 2 years of age, to have lower graft survival rates than older children has been reversed. Some studies even suggest that adult kidneys transplanted into infants, with immediate graft function, may have the longest half-lives of any type of kidney transplant. Pediatric recipients younger than age 11 years who received living donor transplants now have 5-year graft survival rates that are similar to those of recipients in older age groups. The rates were 96% for infants younger than 1 year, 91% for children 1 to 5 years old, and 85% for children 6 to 10 years old. The results for deceased donor recipients are also better in this age group than in adults generally. Recipients 1 to 5 years of age have a 5-year graft survival rate of 75% and recipients 6 to 10 years of age have a 5-year graft survival rate of 72%.
On the other hand, the long-term graft survival rates in adolescents are not as good as those seen in younger children, even though the short-term outcome is similar. The 1- and 5-year graft survival rates for adolescent recipients of living donor kidneys are 94% and 77%, respectively. For deceased donor kidneys, the graft outcomes were 93% and 63%, respectively. Adolescents have the poorest 5-year results of any age group except for recipients 65 years and older. Higher rates of medication noncompliance, loss of medical insurance during transition to adulthood, and a high recurrence rate of FSGS, which is the most common acquired cause of ESRD in this age group, have all been cited as potential causes for the reduced long-term outcome.
Donor Age
For all deceased donor recipients, kidneys from donors 11 to 17 years of age provide optimal graft survival and function. This group is followed next by donors 18 to 34, 6 to 10, and then 35 to 49 years of age. Grafts from donors younger than 5 years fare more poorly, and grafts from donors older than 50 years fare most poorly. Although transplanted kidneys grow in size with the growth of the recipient, transplantation with deceased donor kidneys from donors younger than 6 years is associated with decreased graft survival. The 5-year graft survival rate for recipients of deceased donor kidneys from donors younger than 1 year of age is only about 59%, compared with 73% and 67% for recipients of grafts from donors 1 to 5 years of age and older than 6 years of age, respectively. Kidneys from donors 11 to 17 years of age have the best 5-year graft survival of about 75%. Children younger than 5 years receiving a kidney from a donor younger than 1 year have the highest relative risk for graft failure.
Ethnicity
African American ethnicity is associated with a worse outcome. Five years after transplantation, African American children have graft outcomes of 57% and 70% for recipients of deceased donor and living related donor kidneys, respectively. For white and Hispanic recipients, graft survival rates at 5 years are 74% and 66%, respectively, for recipients of deceased donor kidneys, and 84% for living donor grafts. African American children have not only poorer graft survival but also poorer renal function compared to other ethnic groups.
HLA Matching in Children
In pediatric transplantation, most living donor transplants come from parents. Long-term graft survival is best when the donor is a HLA-identical sibling. When considering transplants from HLA haplotype-identical sibling donors, some studies suggest that there is improved outcome when donor and recipient share “noninherited maternal antigens,” as distinct from “noninherited paternal antigens” (see Chapter 3). Additionally, improved outcomes have been reported with sharing of HLA-DR antigens in living donor transplantation and sharing of HLA-B antigens in deceased donor transplantation, although the differences, if any, are small.
Presensitization
Repeated blood transfusions expose the recipient to a wide range of HLA antigens and may result in sensitization to these antigens, leading to higher rates of rejection and graft failures. The graft failure rate increases by up to 30% for recipients with more than five blood transfusions before transplantation compared with those who had fewer transfusions. Blood transfusions have become less common since erythropoietin became an integral part of ESRD therapy. Hemoglobin levels in children on dialysis are lower than levels in their adult counterparts, and there is support for more aggressive management of anemia to forestall transfusions. Sensitization may also result from rejection and failure of a previous transplant, and the 5-year graft survival rate for repeat deceased donor transplantations is about 13% lower.
Immunologic Factors
Immunologic parameters in younger children are different from those in adults and older children. Such differences include higher numbers of T and B cells, higher CD4+-to-CD8+ T-cell ratio, and increased blastogenic responses. These differences may account for the increased immune responsiveness to HLA antigens and may be partly responsible for the higher rates of rejection that have been observed in children. With improved understanding and management of immunosuppression in pediatric patients, these higher rates of rejection have been significantly ameliorated.
Technical Factors and Delayed Graft Function
Surgical kidney transplant techniques used in older children are similar to those in adults (see Chapter 8). Placement of the vascular anastomoses depends on the size of the child and the vessels. An extraperitoneal approach is usually accomplished with the venous anastomoses to the common or external iliac vein and the arterial anastomoses to the common or external iliac artery. These vascular anastomoses tend to be more cephalad than for adult transplants.
Small children present difficult operative challenges. The relatively large size of the graft may result in longer anastomosis times, longer ischemia time, and subsequently higher rates of early graft dysfunction. When possible, the transplanted kidney is usually placed in an extraperitoneal location, although with very small children, the placement can be intra-abdominal. The aorta and inferior vena cava are usually used for anastomoses to ensure adequate blood flow, but smaller vessels may be used. Vascular anastomosis may be problematic in a child with a previous hemodialysis access placed in the lower extremities or with a previous kidney transplant. Children should be evaluated thoroughly before transplantation to identify any potential anastomotic difficulties. Unidentified vascular anomalies may lead to prolonged anastomosis times and subsequently higher rates of delayed graft function (DGF) and graft thrombosis.
Occasionally, native kidney nephrectomy is necessary at the time of transplantation. Although this can be done routinely in living donor transplantations in which there is little cold ischemia time, it is preferable to avoid this, when possible, in recipients of deceased donor transplants. Native nephrectomy at the time of transplantation prolongs the surgical procedure and may complicate fluid management and contribute to an increase in DGF.
DGF is discussed in detail in Chapter 9. It occurs in about 5% of living donor and 17% of deceased donor transplants and is associated with a reduced graft survival. In children with DGF, the 3-year graft survival rates are reduced by up 25%. Risk factors for DGF in children are more than five prior blood transfusions, prior transplantation, native nephrectomy, African American ethnicity, and a cold ischemia time of longer than 24 hours.
Antibody Induction
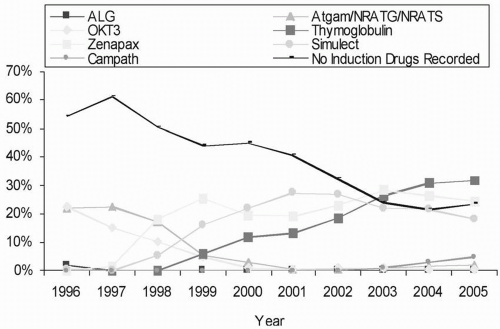
Antibody induction, with either polyclonal or monoclonal antibodies, is used either for prophylaxis against rejection or in a sequential manner to avoid nephrotoxicity resulting from early use of calcineurin inhibitors (see Chapter 5). The NAPRTCS database shows a nearly 17% reduction in the proportional hazard of graft loss with the use of antibody induction in living related transplantation, but surprisingly no significant survival advantage for deceased donor transplantation. Nevertheless, the use of induction agents continues to increase year by year. As a class, nondepleting antibodies (see Chapter 5) remain the most commonly used induction agents for pediatric transplants in the United States, although there has been a steady increase in the use of rabbit antithymocyte globulin (Fig. 16.1).
Transplantation Center Volume
Transplant outcome in high-volume pediatric renal transplantation centers has been reported to be superior to that found in lower-volume centers. High-volume centers (defined as the performance of more than 100 pediatric transplantations between 1987 and 1995) reported a lower incidence of graft thrombosis and DGF, improved long-term graft survival, and more frequent use of antibody induction.
FIGURE 16.1 Immunosuppression use for induction for pediatric recipients with kidney transplants, 1996 to 2005.
Cohort Year
The results of pediatric renal transplantation have been steadily improving. The current 1-year and 5-year graft survival data represent up to 15% improvement during the past 20 years. Graft outcome in transplants from deceased donors performed between 1995 and 2006 is now equivalent to the graft survival in living donor transplantation performed between 1987 and 1994.
Recurrent Renal Disease in Pediatric Transplantation
Recurrent disease in the renal graft accounts for graft loss in almost 8% of primary transplantations and 10% of repeat transplantations. This is more than double that reported for adult transplantation. Both glomerular and metabolic diseases can recur, with most recurrences caused by glomerular disease.
Glomerular Diseases
Focal and Segmental Glomerulosclerosis
FSGS is the most common cause of graft loss as a result of recurrent disease. For patients whose original disease was steroid-resistant nephrotic syndrome or confirmed FSGS, the disease recurs in 30% to 40% of patients undergoing primary transplantation. When the first transplant was lost to recurrence, FSGS recurs in 70% to 85% of those undergoing subsequent transplantation. About 20% to 30% of transplants in patients with the diagnosis of FSGS fail because of recurrence. The mean time to graft failure from recurrence is 17 months.
Recurrence is usually characterized by massive proteinuria, hypoalbuminemia, and nephrotic syndrome with edema or anasarca and hypercholesterolemia. It may present immediately or weeks to months after transplantation. Predictors of recurrence include rapid progression to ESRD from the time of initial diagnosis (<3 years), poor response to therapy, younger age at diagnosis (but older than 6 years of age), Caucasian ethnicity, and the presence of mesangial proliferation in the native kidney. A protein permeability factor has been isolated from sera of patients with FSGS, and its concentration was found to correlate with recurrence and severity of disease in the transplanted kidney. The precise nature of this factor remains unclear, and there is no clinically approved assay to detect it.
Early post-transplantation recognition of recurrent FSGS is important because plasmapheresis (which may lower the serum levels of protein permeability factor) and high-dose calcineurin inhibitor may lead to significant reduction in graft losses because of recurrence. In vitro studies using rat glomeruli show that cyclosporine or tacrolimus, incubated with sera from FSGS patients, will inhibit the proteinuric effect of such sera. Thrice-daily cyclosporine administration may be used in doses that maintain high blood levels (see Chapter 5), and the dose is tapered slowly after achieving remission of the nephrotic syndrome and as cholesterol concentration decreases, or if significant toxicity develops. Some centers have used a high-dose continuous intravenous infusion of cyclosporine with similar improvement, or have used high-dose or thrice-daily administration of tacrolimus. Cyclophosphamide has also been reported to induce remission. Rituximab may prevent recurrence; however, results have been mixed, and its use is more successful in children than adults. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are used as adjuncts to decrease proteinuria. Plasmapheresis is generally used with a frequency that matches disease severity and is occasionally required on a weekly basis for prolonged periods. Plasmapheresis, in combination with a high-dose calcineurin inhibitor, is reported to be superior to either when given alone. Although there is currently no consensus on the treatment regimen for FSGS, the protocol outlined in Table 16.2 represents a summation of our own experience. For deceased donor transplantation, recurrence is less in patients on high-dose cyclosporine and post-transplantation plasmapheresis. For living related transplantation, high-dose tacrolimus is effective when combined with more than five plasma exchanges.
Some studies report that living related donor transplant recipients suffer from a higher rate of recurrence. The graft outcome in recipients of living donor grafts with FSGS recurrence is no better than the outcome observed in recipients of deceased donor grafts that have not experienced recurrence. These data have led some pediatric transplantation centers to reduce or discontinue the use of living related donation for patients with FSGS. However, the controlled settings of living donor transplantation may allow certain benefits in the event that FSGS does recur. The lower incidence of DGF in living donation may permit augmentation of the calcineurin inhibitor dose. In addition, the preplanning implicit in living donation permits preoperative and early postoperative plasmapheresis, an approach that may potentially prevent or decrease the severity of recurrent disease.
Alport Syndrome
Alport syndrome, or hereditary glomerulonephritis, is a progressive disease often associated with neurosensory hearing loss and ocular abnormalities such as anterior lenticonus and cataracts. The inheritance pattern can be X-linked, autosomal recessive, and autosomal dominant. The abnormality in almost all patients stems from mutations in the α3, α4, or α5 helices of type IV collagen. In more than 80% of patients, Alport syndrome results from mutations in the COL4A5 gene on the X chromosome.
Strictly speaking, Alport syndrome itself does not recur; however, antiglomerular basement membrane (anti-GBM) glomerulonephritis occurs in about 3% to 4% of patients after transplantation and can lead to graft loss. The antibodies causing the anti-GBM nephritis are usually directed against the α5 chain of the noncollagenous portion of type IV collagen in the GBM, but antibodies against the α3 chain have also been described. The risk appears to be greatest in patients with mutations of COL4A5 that prevent synthesis of the α5 chain.
TABLE 16.2 Focal Segmental Glomerulosclerosis Protocol at the Mattel Children’s Hospital at UCLA
Identify the high-risk patient
Living related donation if possible (to allow pretreatment and to avoid acute tubular necrosis so that high-dose cyclosporine or tacrolimus can be used)
All patients on angiotensin-converting enzyme inhibitors or angiotensin receptor blockers as tolerated
Living donor graft recipients
○
Ten pretransplantation plasma exchanges (1.5 volumes with albumin; fresh-frozen plasma for patients who are coagulopathic)
○
Three post-transplantation plasma exchanges (may need to be extended)
○
Tacrolimus 2 or 3 times daily; aim for trough levels of 12 to 15 ng/mL
Deceased donor graft recipients
○
Ten post-transplantation plasma exchanges
○
May need to extend the number of plasma exchanges
○
Cyclosporine 3 times daily; aim for trough levels of 200 to 500 ng/mL
Anti-GBM glomerulonephritis presents as rapidly progressive crescentic glomerulonephritis with linear deposits of immunoglobulin G (IgG) along the basement membrane and commonly leads to graft loss, with rates approaching 90%. It usually occurs within the first post-transplantation year. Asymptomatic cases with linear IgG deposits have also been reported. Treatment consists of plasmapheresis and cyclophosphamide, but such treatment is of only limited benefit. Retransplantation is associated with a high recurrence rate.
Membranoproliferative Glomerulonephritis
Histologic evidence of recurrence of membranoproliferative glomerulonephritis (MPGN) type I varies widely, with reported rates ranging from 20% to 50%. Clinical manifestations include proteinuria and deterioration of renal function. Risk factors for recurrence have been reported to include HLA-B8DR3, living related donation, and previous graft loss from recurrence. Graft loss occurs in up to 30% of cases. Histologic recurrence of type II disease occurs in virtually all cases, but graft loss is not inevitable and has been reported in up to 50% of patients. The presence of crescents in the native kidney biopsy and persistent proteinuria may predict severe recurrence that often leads to graft loss. There is no proven treatment for recurrence of MPGN in children. Anecdotal case reports describe success with high-dose corticosteroids, mycophenolate mofetil (MMF), or plasma exchange.
IgA Nephropathy and Henoch-Schönlein Purpura
Histologic recurrence with mesangial IgA deposits is common and occurs in up to 60% of patients with IgA nephropathy and in 35% of patients with HenochSchönlein purpura (HSP). Most of the recurrences are asymptomatic, but graft loss may occur, often associated with crescent formation. Data from adult centers suggest that a fulminant presentation of IgA nephropathy as the original cause of ESRD predicts poor outcome in the transplanted kidney with disease recurrence. In children, up to 10% of graft failures have been ascribed to recurrent IgA nephropathy or HSP. There is no effective therapy for the prevention or treatment of recurrent IgA nephropathy. Anecdotal reports that MMF administration prevents progression to graft failure have not been substantiated. ACE inhibitors and ARBs can be used for reducing proteinuria and preserving renal function.
Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) accounts for up to 5% of primary renal disease in children leading to ESRD. In children, the most frequent form of HUS is diarrhea-associated (D+), or “typical,” and is caused by verocytotoxin-producing Escherichia coli (VTEC). Although this is the most common form of HUS in childhood, it results in ESRD in only 5% to 10% of cases. “Atypical” HUS is far less common in children. It is characterized by a prodrome that lacks diarrheal association (i.e., “D-”); it has a relapsing course and a very poor renal prognosis.
When considering transplantation in patients whose original cause of ESRD was HUS, care must be directed to the form of HUS that the patient suffered. The diarrhea-associated, or typical, form does not usually recur after transplantation, whereas atypical HUS has a high propensity for recurrence. However, there are pitfalls in assessing recurrence of HUS. The D+/D− terminology can sometimes be misleading. Occasionally, patients with VTEC-associated HUS do not have diarrhea and therefore may be mistakenly labeled as D−. Similarly, diarrhea disease can trigger HUS in a patient who is genetically predisposed to HUS and therefore erroneously is characterized as D+ HUS. In addition, it may be difficult to distinguish antibody-mediated vascular rejection from recurrent HUS, which presents histologically as thrombotic microangiopathy (TMA) (see Chapter 14). The calcineurin inhibitors may cause TMA in the transplanted kidney and produce a clinical picture that resembles HUS (see Chapters 5 and 9). Finally, other rarer causes of HUS in the post-transplantation patient may include use of valacyclovir; viral infections such as parvovirus, HIV and cytomegalovirus (CMV); and antibodies against the von Willebrand factor-cleaving metalloproteinase ADAMTS13. With these reservations in mind, it is reasonable to conclude that D+ HUS has a minimal recurrence rate, whereas the aggregate recurrence rate in D− HUS is 20% to 25%. The use of calcineurin inhibitors in both D− and D+ patients does not appear to trigger HUS recurrence, and avoidance of calcineurin inhibitors does not appear to prevent recurrence.
It has been recommended that at least 1 year of clinical quiescence occur before transplantation is attempted for patients with D− HUS, although this hiatus may not reduce the risk for recurrence. The prognosis is poor, with a 10% mortality rate and greater than 80% graft loss rate. For patients who have experienced recurrence, it is estimated that HUS will recur in about 50% of subsequent grafts.
Abnormal complement dysregulation has been associated with a severe form of D− HUS resulting in 60% disease recurrence and more than 90% graft failure. Patients have genetic defects in factor H, factor I, membrane cofactor protein (MCP), factor B, and C3. Pretransplantation genotyping for these mutations is recommended. Factor H deficiency or mutations that impair its function result in a state of continued complement activation with resulting low C3 and C4 levels. High-dose fresh-frozen plasma with plasma exchange has been advocated for this condition. Combined liver-kidney transplantation has been performed in a limited number of patients to restore normal circulating factor H with varying results.
An acquired factor H deficiency as a result of anti-factor H autoantibodies has been found in some children. Plasma exchange can be used to remove anti-factor H antibodies and immunosuppression with steroids and rituximab used to suppress further antibody production. Eculizumab, an anti-C5 monoclonal antibody, has successfully been used to treat D− HUS recurrence by decreasing the damage associated with anaphylatoxin C5a and preventing formation of membrane attack complex on cell surfaces. Randomized control trials of Eculizumab are in progress. Defects in factor I result in impaired protection of the endothelial surface against complement activation; graft recurrence and survival rates are similar to those associated with factor H mutations. Defects in MCP, a transmembrane complement regulator that is highly expressed in the kidney, are associated with a much lower recurrence rate than defects in factor H or I. Transplantation of a kidney that expresses normal MCP should correct the defect. Defects in Factor B, which increase C3bBb convertase stability and result in permanent activation of the complement pathway, have been identified: there is little experience with their clinical management.
Living donor transplantation is not contraindicated for patients whose original disease was D+ HUS. On the other hand, living donor transplantation is not advocated for patients with D− HUS. This is because of the high recurrence rate in such patients. In addition, it has been noted that some parental carriers of D− HUS might not manifest the disease until later in life, and organ donation would put such carriers at excessive risk.
Antiglomerular Basement Membrane Disease
Anti-GBM disease is rare in children. A high level of circulating anti-GBM antibody before transplantation is thought to be associated with higher rate of recurrence. Therefore, a waiting period of 6 to 12 months with an undetectable titer of anti-GBM antibody is recommended before transplantation to prevent recurrence. Reappearance of anti-GBM antibody in the serum may be associated with histologic recurrence. Histologic recurrence has been reported in up to half of cases, with clinical manifestations of nephritis in only 25% of these cases. Treatment for recurrence includes plasma exchange, cyclophosphamide, and corticosteroids. Graft loss is rare, and spontaneous resolution may occur.
Congenital Nephrotic Syndrome
Congenital nephrotic syndrome occurs in the first 3 months of life. It can be classified by mutations in the nephrin gene (NPHS1), podocin gene (NPHS2), or Wilms tumor suppressor gene (WT1). Congenital nephrotic syndrome of the Finnish type (CNSF) is an autosomal recessive disease that occurs as a result of a mutation in the NPHS1 gene. Although it is most commonly seen in Finnish patients, it is also found in other countries. The NPHS1 gene is located on chromosome 19 and has as its gene product the protein nephrin. Nephrin is a transmembrane protein, which is a member of the immunoglobulin family of cell adhesion molecules. It is characteristically located at the slit diaphragms of the glomerular epithelial foot processes. Close to 100 mutations of NPHS1 have been identified in CNSF, but more than 90% of all Finnish patients have one of two mutations—the so-called Fin major and Fin minor mutations.
Infants with CNSF are usually born prematurely and exhibit low birth weight and placentomegaly. CNSF manifests as heavy proteinuria, edema, and ascites, often in the first week of life and always by 3 months of age. Renal histology is nonspecific and shows expansion of glomerular mesangium and dilations in the proximal and distal tubules. Untreated, these children suffer from malnutrition, poor growth, frequent infections, and thromboembolic complications. ESRD occurs invariably by mid-childhood. Corticosteroids do not ameliorate CNSF, but in mild forms, ACE inhibition, together with indomethacin, may be successful. The best therapeutic success has come from the approach of early dialysis, nephrectomy, and transplantation.
CNSF does not recur after transplantation. However, de novo nephrotic syndrome has been reported in about 25% of cases. It presents with proteinuria, hypoalbuminemia, and edema that may start immediately or as late as 3 years after transplantation. All the patients with post-transplantation nephrotic syndrome have been reported to have the homozygous Fin major genotype. Antibodies against fetal glomerular structures are found in most patients with post-transplantation nephrotic syndrome, and antibodies to nephrin are found in more than 50%. About half of patients with this nephrotic syndrome respond to steroids and cyclophosphamide, but in those who do not respond, the graft is usually lost. Plasma exchange to decrease antinephrin antibodies has been a useful adjunct. In the NAPRTCS database, vascular thrombosis and death with a functioning graft (mostly as a consequence of infectious complications) occur in 26% and 29% of cases, respectively, and account for a higher rate of graft failure in this particular group.
Mutations in the NPHS2 gene located on chromosome 1 account for half of the congenital nephrotic syndrome cases in 80 European families and are autosomal recessive. Podocin is a podocyte-adapter protein required for proper targeting of nephrin into the slit diaphragm. Patients who are homozygous for podocin mutations develop early-onset steroid-resistant nephrotic syndrome, usually in infancy or early childhood, and usually progress to ESRD. Renal histology shows FSGS. Because podocin is a structural component of the glomerular filtration barrier, it was hypothesized that deficient podocin was the cause of renal disease and that recurrence would not occur. However, there are reports of recurrence in recipients from parents who are obligate carriers of NPHS2 and in patients with heterozygous NPHS2 mutations. The mechanisms remain unclear. Response to plasma exchange has been variable.
Mutations in WT1 gene located on chromosome 11pl3 account for some cases of congenital nephrotic syndrome. WT1 transcription factor plays a crucial role in the embryonic development of the kidney and genitalia. It is abundantly expressed in podocytes and controls cellular functions, such as nephrin expression. Patients with WT1 mutations have moderate proteinuria, and renal biopsy reveals diffuse mesangial sclerosis (DMS) of glomeruli. WT1
Only gold members can continue reading. Log In or Register to continue