Fig. 14.1
Hypertension induces inflammatory processes. Hypertension can promote inflammatory events within the vascular wall and the kidney. Increased sympathetic nerve activity (SNA) can drive both hypertension and immune cells activation. EC endothelial cells, SMC smooth muscle cells, ROS reactive oxygen species, Aldo aldosterone, AngII angiotensin II
Accumulating evidence from basic science research and clinical studies extensively demonstrated that AngII, besides regulating the vascular tone, could exert pro-inflammatory effects within the arterial wall and other organs. AngII, in fact, induces NF-kB activation by triggering the production of inflammatory cytokines , promotes the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is followed by the release of ROS, and impairs endothelium-dependent vasodilatation by reducing nitric oxide (NO) generation [24]. Some of the hypertensive effects of AngII can be also driven by the modulation of the immune/inflammatory response. For example, it has been demonstrated in mice that lack the production of IL-17 (IL-17(−/−)) that the rise in blood pressure induced by AngII infusion was less sustained compared to wild-type animals. Moreover, the vessels of IL-17(−/−) mice showed preserved vascular function, lower superoxide production, and less T cell infiltration within the arterial wall as compared to control animals [25]. As mentioned above, inflammatory processes including immune cell recruitment, cytokine release, and ROS production are commonly observed within the kidneys of experimental model of hypertension . These processes can actively contribute to local tissue damage and induce progressive impairment of renal hemodynamic and tubular function [26]. In this context, a central role can be played by intrarenal AngII, whose expression has been found to be correlated with the severity of hypertension and the level of immune cell infiltration in the kidney [27]. In fact, locally activated renin–angiotensin system (RAS) can contribute to the pathogenesis of renal damage, thus driving the recruitment and inflammatory activation of immune cells [28]. In addition, treatment of animal models of hypertension with RAS blockade have been shown to reduce the renal inflammatory infiltration mainly through reduction of chemokines, cytokines, and adhesion molecules expression [28]. Similar effects have been observed within the arterial wall, where the RAS inhibition was able to reverse most of the detrimental effects of AngII on endothelial function and to reduce the level of inflammatory activation in the vessels [20, 24]. This basic science data found some confirmation in clinical studies showing that treatment with AT-1 receptor blockers can lower the circulating levels of some inflammatory mediators (such as IL-6, TNFα, MCP-1, hs-CRP, and CD40L) [29–31]. Also, aldosterone , an important component of the RAS system, has been shown to actively contribute to vascular and kidney damage, by inducing oxidative stress, endothelial dysfunction, fibrosis, and inflammation [32]. In addition, aldosterone inhibits NO synthase thereby reducing NO production both within the kidney and vessels [33]. As for AngII, also the pharmacological blockade of aldosterone has been proved to reduce pro-inflammatory and pro-fibrotic vascular damage independently from the effects on blood pressure. For instance, despite similar blood pressure reduction, treatment with mineralcorticoid receptor (MR) blockade resulted more efficiently than treatment with atenolol in reducing vascular stiffness and systemic inflammation among hypertensive subjects [34]. Of interest, the expression of MR has also been demonstrated in immune cells (mainly monocytes/macrophages) suggesting a role for aldosterone in promoting vascular damage through inflammatory activation of this cell type. In addition, it has been shown that macrophages, through MR receptor, could also mediate some of the hypertensive effects of aldosterone. In fact, data obtained in the deoxycorticosterone acetate (DOCA)-salt-hypertension mouse model demonstrated that the selective lack of MR expression in monocytes is followed by no increase in either cardiac fibrosis or blood pressure level [35]. Several pieces of evidence arising from animal models and clinical studies also showed that MR blockade with either eplerenone or spironolactone is accompanied by reduction in renal inflammation, oxidative stress, proteinuria, and glomerular and tubular injury [36]. Nevertheless, the safety as well as the renal and CV outcomes of treatment with low-dose MR antagonists of CKD patients is still uncertain, and large prospective studies are needed to clarify this aspect.
Mechanical stress and humoral factors are also considered as important stimuli for the activation of medial and adventitial vascular cells. In this context, a pivotal role is played by vascular SMC, which harbor remarkable plasticity in terms of differentiation, proliferation, and motility. We and others observed that specific immature type of SMC populations can be found within the arterial wall, and that these cells are actively involved in the vascular remodeling associated with hypertension [37]. Both medial SMC and adventitial myofibroblasts exposed to mechanical forces and growth factors could undergo a phenotypic dedifferentiation towards the acquisition of a “synthetic” profile [38]. Following this transition the vascular cells exhibit an increase in ability to migrate towards the intima layer, higher secretion of inflammatory mediators, and higher capacity of extracellular matrix (ECM) remodeling [38, 39]. These cellular modifications are influenced both by hemodynamic and bioumoral factors (including ROS, AngII, and aldosterone) and lead to the generation of pro-fibrotic vascular damage. In addition, together with endothelial dysfunction and inflammatory cells recruitment, vascular cells proliferation/migration within the neointima represent initial steps of atherogenesis and certainly represent a pathophysiological connection between hypertension and atherosclerosis development. The amplification of the inflammatory response associated with the hypertensive damage of the arteries could increase the circulating levels of inflammatory molecules, and partly accounts for the low-grade inflammatory status observed in hypertensive subjects.
It is well established that the sympathetic nerve activity (SNA) plays a crucial role in the regulation of blood pressure mainly through the modulation of peripheral arterial tone and cardiac output. In particular, data in humans suggest that a neurogenic component can be observed in 40–65 % of hypertensive patients [40]. Growing evidence suggest that SNA could significantly interplay with the immune system. Lymphoid organs are highly innervated by the sympathetic nerves and stress hormones such as catecholamines have been shown to possess immunomodulatory properties [41]. In particular, early studies suggested that stress hormones may exert immunosuppressive effects through the inhibition of the T helper lymphocyte 1 (Th1) pro-inflammatory activities, as well as the induction of Th2 anti-inflammatory cytokines production. However, more recent findings indicate that in certain conditions, the hyperactive stress system might instead exert pro-inflammatory effects, thus influencing the onset of several human immune-related diseases, including vascular disease progression [41]. Cerebral infusion of AngII has been shown to increase SNA, which in turn increases the expression of inflammatory cytokines (such as IL-1, IL-2, IL-6, IL-16) in splenocytes. This inflammatory activation of the cells was blunted by splenic sympathectomy [42]. Data are available showing that immune cells possess both adrenergic and cholinergic receptors and that these receptors significantly impact on their function. For example, T cells possess both α- and β-adrenergic receptors which have been shown to be actively involved in cell proliferation, Th1/Th2 polarization, and change of surface markers [43]. In addition, expression of α1- and β1-adrenergic receptors have been demonstrated on the surface of monocytes and macrophages and its stimulation with adrenergic receptor agonists significantly enhance the pro-inflammatory cytokine production by the cells in response to toll-like receptor (TLR) agonists [44, 45]. Additional investigations showed that monocyte and macrophages express α7-nicotinic receptors and its activation suppress cytokine production [46, 47]. Thus, it is plausible that an increase in SNA often observed in hypertensive patients might contribute to the overall low-grade inflammatory state also through its immunomodulatory effects. On the other hand, the immune system could represent an important mediator of SNA contribution of hypertension.
Does Inflammation Promote Hypertension?
Evidence so far collected does not clearly establish whether or not inflammation per se could induce structural/functional changes of the arterial wall which lead to the development of hypertension . Nevertheless, several lines of research on this topic have been developed in the past few years and novel theories are now under intense investigation (Fig. 14.2). As a first hypothesis, it could be postulated that, independently from their source, inflammatory circulating molecules might significantly impact on mechanisms of arterial tone regulation. In particular, it has been shown that inflammatory molecules (such as IL-6, IL-1, TNFα, CRP, and others) can produce detrimental effects on the vascular wall, including EC dysfunction with reduced NO bioavailability, an increased expression of adhesion molecules, and release of ROS [48]. Some of these cytokines have been also shown to mediate AngII hypertensive and pro-atherogenic effects. For instance, in mice it has been shown that IL-6 deficiency protects against AngII-induced increases in superoxide, EC dysfunction, and vascular hypertrophy [49]. Of note, the knockdown of IL-6 was also able to blunt the AngII-driven rise in blood pressure [50]. A similar effect and interaction with AngII have also been described for TNFα. In particular, it has been shown that the use of etanercept, a TNFα antagonist, prevented hypertension induced by fructose feeding and AngII infusion [51, 52]. Both IL-6 and TNFα together with other cytokines are increased in the kidneys of animal models of hypertension, suggesting that they can be important mediators in the link between renal injury and blood pressure elevation. In line with this possibility, it has been demonstrated that inhibition of both TNFα (with etanercept) and IL-6 (with small hairpin RNA) attenuated both the inflammatory damage of the kidney and the development of hypertension [53, 54]. Together with pro-inflammatory cytokines some studies have been conducted to dissect the role played by anti-inflammatory cytokines (such as IL-10) during hypertension development. Of interest, data obtained from IL-10-deficient mice showed that this cytokine is able to prevent the vascular dysfunction induced by AngII [55]. Moreover, it has also been observed that IL-10 infusion was able to reduce blood pressure, endothelial dysfunction, and urinary protein excretion in a pregnancy hypertension model [56]. Also, chemokines, important mediators of immune cells infiltration within the vessels, can be actively involved in the pathogenesis of hypertension. For instance, data are available showing that pharmacological blockade of chemotactic cytokine receptor-2 (CCR2; main receptor of the chemokine monocyte chemoattractant protein-1, MCP-1) attenuated both macrophages aortic infiltration and hypertension development in the DOCA-salt animal model [57]. Some of these pro-hypertensive effects of cytokines and chemokines could also be ascribed to their role in modulating proliferation, migration, and synthetic behavior of vascular SMC [58]. In fact, evidence available shows that exposure of vascular SMC to IL-6, TNFα, CRP, and other cytokines is followed by cellular inflammatory activation with increase proliferation/migration and enhanced capacity of ECM remodeling (mainly through the secretion of metalloproteinases and collagen synthesis) [59, 60]. As mentioned above, these pathological processes are mainly characterized by the involvement of SMC harboring an immature/synthetic phenotype. We previously observed that arterioles obtained from hypertensive patients displayed higher amount of immature SMC compared to those obtained from normotensive subjects [61]. Additional investigations are needed to establish whether, besides modulating SMC phenotype during atherogenesis, inflammatory mediators can also impact on the morphological and functional behavior of SMC resident within the resistance arterioles.
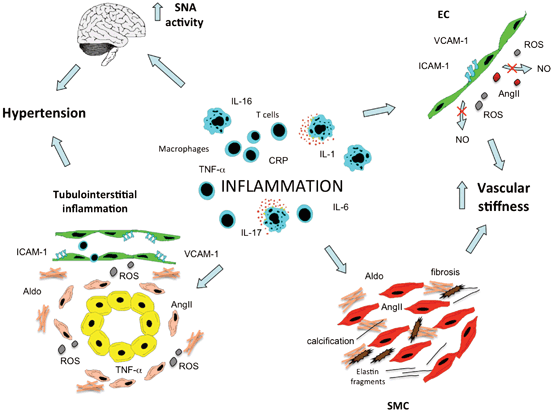
Fig. 14.2
Inflammation can induce hypertension. Inflammatory processes within the arterial wall can induce endothelial cell (EC) dysfunction and smooth muscle cell (SMC) activation, which both lead to increase of vascular stiffness. Moreover, also inflammatory events involving the kidney and the central nervous system can promote hypertension. SNA sympathetic nerve activity, ROS reactive oxygen species, Aldo aldosterone, AngII angiotensin II, NO nitric oxide
Inflammatory activation of both EC and SMC can also be associated with significant changes in large vessels structure, mainly increase in arterial stiffness and reduction in vascular resilience. The rise in SBP, the fall in DBP and the resulting increase in pulse pressure are now considered as a manifestation of the presence of central arterial stiffening. The development of this blood pressure pattern, which is commonly observed with aging, is accelerated among subjects with advanced stages of CKD and diabetes. Moreover, several epidemiological investigations demonstrated the negative prognostic value of arterial stiffness for future CV events [62]. Stiffening of the arteries is mainly characterized by fracturing of elastin together with increase of collagen and calcium accumulation. Traditional view interprets reduced elasticity of the arteries as a consequence of hypertension. However, some recent studies underline the possibility that arterial stiffening might not only precede the development of hypertension [63] but also drive, at the later stages, the development of systolic hypertension [64]. Of note, pulse wave velocity (PWV), a measure of large vessels stiffness, has been directly associated with the circulating levels of some inflammatory mediators (such as CRP, IL-6, and TNFα) [65–67] suggesting that inflammation may contribute to arterial stiffness, maybe through induction of EC dysfunction and SMC activation. Vascular calcification is now identified as a major determinant of arterial stiffening especially among patients with advanced stages of CKD, and its presence has been linked to the increased CV mortality observed in this group of subjects [68]. Growing evidence suggested that calcium accumulation within the arterial walls and the heart valve leaflets cannot be considered just a passive phenomenon [64, 69, 70]. Osteoblast/chondrocytes-like cells have been identified in vivo within pathological vascular tissue of humans and animal models of atherosclerosis. In addition, the expression of osteogenic markers has been shown to significantly increase within the arteries of patients with advanced stages of renal failure [71]. A number of in vitro experiments demonstrated that both valve cells and vascular SMC exposed to inorganic phosphate [72], uremic factors, and inflammatory mediators (such as TNFα and IL-6) [73] can acquire an osteogenic phenotype and deposit calcium [74]. More recently, AngII has been implicated in the pro-calcific differentiation of vascular cells and its effect seems to be driven by the induction of receptor activator of nuclear factor kappa-B ligand (RANKL) which prevents the anti-calcific effects of osteoprotegerin [75]. It is plausible that all these humoral and inflammatory factors can synergically participate within the pathological vessel in promoting elastin damage, collagen/calcium deposition and thus leading to arterial stiffness. The latter is then associated with worsening of blood pressure levels and favors the onset of future CV events.
Monocytes/macrophages are known to be major contributors to cytokines/chemokines release during vascular and kidney disease progression. These cells are also the source of ROS, metalloproteinases, and other factors that induce tissue damage and promote vascular remodeling which in turn leads to increased vasoconstriction and arterial stiffness through the mechanisms listed above. Recent data showed that selective deletion of circulating monocytes is able to attenuate AngII-induced blood pressure elevation and vascular dysfunction, including the induction of vascular adhesion molecules and ROS generation [76].
Growing evidence collected in the past few years underscored that beside the role played by elements belonging to the innate immunity response (such as cytokines, chemokines, and monocytes/macrophages), components of the adaptive immunity might also be actively involved in the pathogenesis of hypertension. Early data obtained from animal models showed that transfer of lymph node cells, splenocytes, and lymphocytes obtained from hypertensive rats were able to raise the blood pressure in the normotensive recipients [77–79]. Moreover, the removal of thymus was followed by blood pressure reduction in rats with renal hypertension [80]. More recently, it has been clearly shown that lymphocytes play a causative role in the rise of blood pressure observed in different animal models [4]. For instance, AngII-induced blood pressure rise was significantly blunted in RAG-1-deficient mice, which lacks both T and B cells. These animals also displayed a significant reduction of vascular dysfunction induced by AngII, including ROS production. Of interest, only the adoptive transfer of T lymphocytes, and not the B cells, was able to restore the AngII-induced hypertension [51]. Similar findings were confirmed by using other models of animal hypertension (such as the norepinephrine and DOCA-salt mice) [81] and immunodeficiency (such as the severe combined immunodeficiency (SCID) mice) [82]. An opposite, protective effect has been instead recently proposed for regulatory T cells (Tregs), an immunosuppressant subset of T cells , that are thought to be protective against vascular disease progression. Interestingly, it has been shown that Tregs are reduced in the renal cortex during AngII-induced hypertension and that adoptive transfer of these cells is able to ameliorate AngII effects on blood pressure, renal inflammation, and cardiac damage [83, 84].
As mentioned above, evidence obtained from animal models of hypertension showed that an inflammatory infiltration by immune cells occurs in the kidney. Of interest, this renal tubulointerstitial inflammation is observed at a young age of the animals and seems to precede the onset of hypertension [85], thus suggesting a causative role of kidney inflammation in hypertension generation. Early studies showed that the immune cells recruitment in the kidney can be prevented by blocking NF-kB activation, a master transcription factor controlling inflammatory responses. Of note, NF-kB inhibition was accompanied by complete abrogation of hypertension development in spontaneously hypertensive rats [86]. In line with this finding, data are available demonstrating that immune cell infiltration is actively involved in the pathogenesis of hypertension in Dahl salt-sensitive rats. In fact, immunosuppressive therapy with mycophenolate mofetil in this animal model was accompanied by reduced T and B recruitment in the kidney, and significantly attenuated the development of hypertension [87, 88]. Further studies also suggested that the detrimental effects of T lymphocytes on hypertension and kidney damage are due to local release of AngII and increased production of ROS [5]. Thus, it could be assumed that the infiltration of immune cells, and the associated increase in inflammatory cytokines and oxidative stress within the renal interstitium, can have a prominent role in the pathogenesis of hypertension. Nevertheless, we do not actually know if this phenomenon has some relevance on humans.
Sympathetic overdrive is actively contributing in generating essential hypertension and in particular the so-called neurogenic hypertension. Interestingly, vascular inflammation affecting the cerebral vasculature and, in particular, the brainstem CV control areas has been recently implicated in the generation of this form of hypertension [89]. In particular, it has been demonstrated that the microvasculature in the nucleus of the solitary tract (NTS) of hypertensive animals expresses higher level of pro-inflammatory molecules (such as JAM-1 and LTB4 12-HD) as compared to normotensive animals [89]. Moreover, overexpression and injection of these molecules in normotensive rats was followed by increased arterial pressure. It is known that RAS is involved, within the NTS, in the CV regulation. Of note, evidence has been collected and shows that the central actions of AngII in modulating SNA are mediated by inflammatory molecules. For instance, it has been shown in rat infused with AngII that the intracerebroventricular administration of minocycline (an anti-inflammatory antibiotic) is followed by a significant reduction of IL-6, IL-1β, TNFα, and an increase of IL-10 in the microglia of the brain. This concomitant attenuation in pro-inflammatory cytokines and increase in anti-inflammatory molecules is followed by significant reduction in blood pressure, cardiac hypertrophy, and plasma levels of norepinephrine [90]. These findings coupled with the observations reported above allow depicting a vicious cycle scenario, with inflammatory mechanisms controlling SNA in the brain, and sympathetic activity modulating immune responses in the periphery with subsequent induction of vascular inflammation/dysfunction.
Conclusions
Data collected from clinical and basic science studies showed a complex mosaic of interplay between local and systemic inflammation, central nervous system, and vascular cells. It appears that effectors of inflammation (immune cells, cytokines, chemokines) and hypertension could interact with each other in a bidirectional manner and that numerous feedback loops probably exist between these two conditions (Fig. 14.3). Of note, the inflammatory processes taking place in the kidney appear to play a central role not only during CKD progression but also in the pathogenesis of systemic hypertension. Further investigations are needed to clearly identify factors initiating these pathological events and the mechanisms driving the interplay between vascular/renal inflammation and sympathetic system activity. Data collected from such studies could offer the opportunity to identify novel therapeutic targets for the treatment of hypertension and its CV complications.
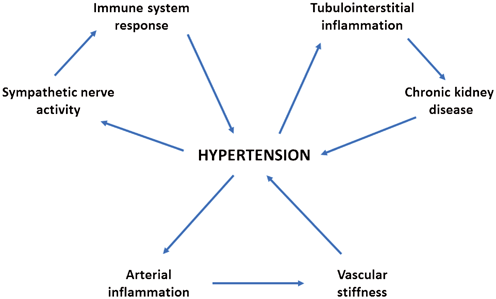
Fig. 14.3
Hypothetical interplay between hypertension and inflammation. Inflammatory processes induced by high blood pressure can be observed both within the kidney and the arteries. The resulting reduction in renal function and increase in vascular stiffness are then followed by further elevation in blood pressure levels. The increase of sympathetic nerve activity that is often observed in hypertensive subjects has been shown to induce pro-inflammatory activation of the immune system. Moreover, both innate and adaptive immune responses can actively contribute in the pathogenesis of hypertension (see the text for details)
References
2.
3.
Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–51.CrossRefPubMed
Related posts:
 Neurogenic Factors in Hypertension Associated With Chronic Kidney Disease
Dual Inhibitors: RAAS Blockers/Combination Therapies: What Do All These Trials Mean?
Genome-Wide Association Studies (Gwas) of Blood Pressure in Different Populations
Neurogenic Factors in Hypertension Associated With Chronic Kidney Disease
Dual Inhibitors: RAAS Blockers/Combination Therapies: What Do All These Trials Mean?
Genome-Wide Association Studies (Gwas) of Blood Pressure in Different Populations
 Resistant Hypertension in Patients with Chronic Kidney Disease
Resistant Hypertension in Patients with Chronic Kidney Disease
 Changes in Guideline Trends and Applications in Practice: JNC 2013 and the Future
Changes in Guideline Trends and Applications in Practice: JNC 2013 and the Future
 Uric Acid and Hypertension: Is There Really a Link?
Uric Acid and Hypertension: Is There Really a Link?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


