Cyclosporine is a small cyclic polypeptide of fungal origin. It consists of 11 amino acids and has a molecular weight of 1203. It is neutral and insoluble in water but soluble in organic solvents and lipids. The amino acids at positions 11, 1, 2, and 3 form the active immunosuppressive site, and the cyclic structure of the drug is necessary for its immunosuppressive effect. Tacrolimus, still often called by its nickname Eff-Kay from its laboratory designation FK506, is a macrolide antibiotic compound isolated from Streptomyces tsukubaensis.
Mechanism of Action
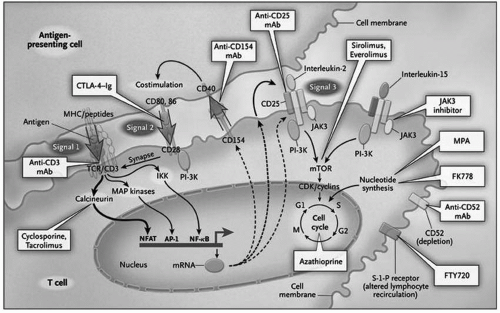
The calcineurin inhibitors differ from their predecessor immunosuppressive drugs by virtue of their selective inhibition of the immune response. They do not inhibit neutrophilic phagocytic activity as corticosteroids do, nor are they myelosuppressive. Cell surface events and antigen recognition also remain intact (see
Chapter 2). Their immunosuppressive effect depends on the formation of a complex with their cytoplasmic receptor proteins,
cyclophilin for cyclosporine and tacrolimus-binding protein (
FKBP) for tacrolimus (
Plate 5.1). This complex binds with
calcineurin, whose normal function is to act as a phosphatase that dephosphorylates certain nuclear regulatory proteins (e.g.,
nuclear factor of activated T cells) and hence facilitates their passage through the nuclear membrane (see
Chapter 2 and
Fig. 2.5). Inhibition of calcineurin thereby impairs the expression of several critical cytokine genes that promote T-cell activation, including those for IL-2, IL-4, interferon-γ (IFN-γ ), and tumor necrosis factor-α (TNF-α). The transcription of other genes, such as CD40 ligand and the proto-oncogenes H-
ras and c
-myc, is also impaired. The importance of these factors in T-cell activation is discussed in more detail in
Chapter 2, but as a result of calcineurin inhibition, there is a quantitative limitation of cytokine production and downstream lymphocyte proliferation.
Cyclosporine enhances the expression of transforming growth factor-β (TGF-β), which also inhibits IL-2 and the generation of cytotoxic T lymphocytes, and may be responsible for the development of interstitial fibrosis, an important feature of calcineurin inhibitor nephrotoxicity. TGF-β has also been implicated as an important factor in the proliferation of tumor cells, which may be relevant to the course of certain post-transplantation neoplasias (see
Chapter 10). The
in vivo effects of cyclosporine are blocked by anti-TGF-β, indicating that TGF-β may be central to the mediation of both the beneficial and detrimental effects of the calcineurin inhibitors.
Patients receiving successful calcineurin inhibitor-based immunosuppression maintain a degree of immune responsiveness that is still sufficient to
maintain host defenses. This relative immunosuppression may be a reflection of the fact that at therapeutic levels of these drugs, calcineurin activity is reduced by only about 50%, permitting strong signals to trigger cytokine expression and generate an effective immune response. In stable patients receiving cyclosporine, CD4+ T cells have reduced IL-2 production to a degree that is inversely correlated to drug levels. The degree of inhibition of calcineurin activity and IL-2 production may be at the fulcrum of the delicate balance that exists between too much and too little immunosuppression.
Formulations and Pharmacokinetics
Cyclosporine. The original formulation of cyclosporine, the oil-based Sandimmune, has largely been replaced by the microemulsion formulation, Neoral. Both formulations are available in two forms: a 100-mg/mL solution that is
drawn up by the patient into a graduated syringe and dispensed into orange juice or milk, and 25-mg and 100-mg soft-gelatin capsules. Patients usually prefer the convenience of the capsule that is typically administered twice daily.
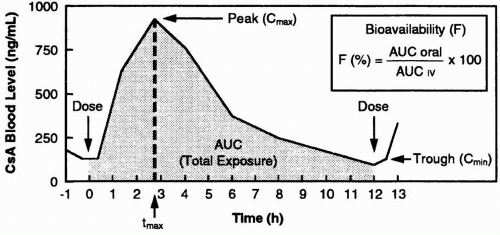
The absorption of cyclosporine after an oral dose can be represented graphically in the form of a concentration-time curve (
Fig 5.1). The time to peak concentration of Sandimmune cyclosporine (
tmax) is variable but averages 4 hours. A substantial proportion of transplant recipients exhibit a second peak. The bioavailability of Neoral (
F) is better than that of Sandimmune, and there is less variability in cyclosporine pharmacokinetics. Peak cyclosporine levels (
Cmax) of Neoral cyclosporine are higher, and the trough concentration (
Cmin) correlates better with the systemic exposure, as reflected by the
area under the curve (AUC).
The improved gastrointestinal (GI) absorption of the microemulsion and lesser dependence on bile for absorption may reduce the necessity for intravenous cyclosporine administration. Compared with intravenous infusion, the bioavailability of the orally administered drug is in the range of 30% to 45%. Conversion between the oral and intravenous forms of the drug perioperatively requires a 3:1 dose ratio. Bioavailability of oral cyclosporine increases with time, possibly as a result of improved absorption by the previously uremic GI tract. As a result, the amount of cyclosporine required to achieve a given blood level tends to fall with time and typically reaches a steady level within 4 to 8 weeks. Food tends to enhance the absorption of cyclosporine (see
Chapter 19).
The development of generic formulations of cyclosporine and other immunosuppressive agents is controversial because of the critical importance of these drugs to the success of transplantation and the corporate and financial implications of their introduction. Cyclosporine is regarded as a drug with a
narrow therapeutic index, and the standards for proving the
bioequivalence of generic forms are more rigorous. Generic drugs, however, do not undergo the same extensive evaluation required of new drugs, and information on discrete differences in their pharmacokinetics in different ethnic groups is not available. Generic formulations of cyclosporine, such as the capsule
cyclosporine USP (Eon Labs) and the capsule
Gengraf, are in widespread use in the United States; other generic formulations are available outside of the United States. The
generic formulations are generally claimed to have an absorption profile that is very similar to that of Neoral. The capsules have received a so-called AB rating by the U.S. Food and Drug Administration (FDA), which means that they may be substituted for Neoral cyclosporine without the approval of the prescriber. If generic formulations are used, it is probably better to use them consistently and to avoid switching formulations. If conversions are made between the different formulations, it is wise to monitor drug levels and renal function (see
Part IV). Extensive experience with generic formulations of cyclosporine has not demonstrated them to be inferior to the brand drug.
Tacrolimus. Tacrolimus (Prograf) is available in an intravenous formulation and as 5-mg, 1-mg, and 0.5-mg capsules. It is typically administered twice daily. A long-acting once-daily formulation (Advagraf) is available in Europe but not in the United states. GI absorption is independent of bile salts. Because of the effectiveness and relative consistency of its absorption, it is rarely necessary to use the intravenous formulation, and if necessary, the drug can be administered through a nasogastric tube. It is absorbed primarily from the small intestine, and its oral bioavailability is about 25%, with large interpatient and intrapatient variability, particularly for patients with GI disease. Gastric emptying of solids is faster in patients taking tacrolimus than in those receiving cyclosporine, a property that may be beneficial for patients with gastric motility disorders. Diarrhea may lead to increased absorption of tacrolimus from the lower GI tract with resultant toxic levels. Generic formulations of tacrolimus are being developed, but as of early 2009, they are not available in the United States. When available, their safety and effectiveness will need to be evaluated with great care.
Drug-Level Monitoring
The measurement of cyclosporine and tacrolimus levels is an intrinsic part of the management of transplant patients because of variation in interpatient and intrapatient metabolism. There is also a relationship, albeit an inconsistent one, between blood levels of the drug and episodes of rejection and toxicity. Drug-level monitoring is the source of much confusion because of the various assays available and the option of using different matrices (i.e., plasma or whole blood) for their measurement.
When Sandimmune was introduced, the trough level of cyclosporine (drawn immediately preceding the next dose), rather than the peak level, was measured because its timing was more consistent and appeared to correlate better with toxic complications. More sophisticated techniques of monitoring were suggested whereby a full, or abbreviated, pharmacokinetic profile is constructed to calculate the AUC, which reflects the bioavailability of the drug and may theoretically allow for more precise and individualized patient management. Although attractive, these techniques never proved popular because of their cost and inconvenience.
Evidence suggests that because of the more consistent absorption of Neoral cyclosporine, its peak level (typically 2 hours after dosing;
Fig. 5.1) may correlate better with drug exposure and clinical events than the trough level. So-called C2 monitoring is applied routinely in some centers and clinical trials. For tacrolimus, the trough level is used for monitoring, and this level is an adequate approximation of drug exposure. Recommendations for target blood levels at different stages after transplantation are discussed in
Part IV.
Cyclosporine concentrations can be measured in plasma or whole blood. Whole blood (ethylenediaminetetraacetic acid [EDTA] anticoagulated) is the recommended specimen type because the distribution of cyclosporine between plasma and erythrocytes is temperature dependent. The clinician cannot begin to assess the significance of a cyclosporine level without knowing what kind of assay is being used. Several methods are currently available to measure cyclosporine, and each differs in specificity for parent compound.
High-performance liquid chromatography (HPLC) is the most specific method for measuring unmetabolized parent cyclosporine and is considered the reference method. HPLC, however, is expensive and labor intensive and is not available at all centers. Immunoassays, which use monoclonal antibodies against cyclosporine, are commonly used and have largely replaced HPLC because they can be performed on automated chemistry analyzers. The most commonly used immunoassay to measure cyclosporine in whole-blood samples is the Abbott (Chicago, IL) fluorescence polarization immunoassay (FPIA), which has significant cross-reactivity with cyclosporine metabolites and overestimates cyclosporine by as much as 45%. Samples for quantitation of peak cyclosporine levels should be clearly identified when sent to the laboratory and should be reported as such. These samples may exceed the linearity of the assay and will need to be diluted for accurate quantitation. For monitoring of tacrolimus concentrations, most laboratories use the Abbott monoclonal antibody-based
microparticle enzyme immunoassay (MEIA) that can be performed on an automated instrument (IMx). This assay permits accurate estimation of tacrolimus levels as low as 2 ng/mL. Abbott has also developed a
chemiluminescent microparticle immunoassay (CMIA) that is available on the
ARCHITECT family of instruments with a reported detection limit of less than 1 ng/mL. Target cyclosporine (peak and trough) and tacrolimus (trough) levels are discussed in the section on immunosuppressive protocols.
Drug Interactions
The interaction of the calcineurin inhibitors with many commonly used drugs demands constant attention to drug regimens and cognizance of potential interactions. New drugs should be introduced with care, and patients should be warned to consult drug package inserts and physicians familiar with the use of cyclosporine and tacrolimus before considering new pharmacologic therapy. Some of the drug interactions discussed below are consistent and well established (and are emphasized in
bold lettering); others have been described in small series and case reports or are anticipated based on the pharmacologic properties of the agents. Any drug that impacts on P-450 activity in the liver or intestinal tract, or that interacts with a drug that does, should be regarded as having a potential interaction with the calcineurin inhibitors. Some drugs affect calcineurin inhibitor levels when administered orally, but not intravenously, because the drug interaction is taking place at the intestine. In addition to their affect on P-450, the calcineurin inhibitors inhibit multidrug resistance protein (MDR), and many of the interactions thought to be due to P-450 are, in fact, due to an effect on MDR. The possibility that the calcineurin inhibitor is affecting the blood level of the interacting drug should also be considered. Unless a comment is made to the contrary, the drug interactions noted below are common to both cyclosporine and tacrolimus, although more have been described with cyclosporine, which has been available longer. Drug interactions between calcineurin inhibitors and other immunosuppressive drugs are discussed in
Part IV. Interactions with antibiotics are discussed below and in
Chapter 11. Interactions with food are discussed in
Chapter 19. Interactions with psychotropic drugs are discussed in more detail in
Chapter 17. Drugs that cause impairment of graft function by virtue of their nephrotoxicity alone are not specifically discussed here.
It should be emphasized that the sections below are not intended to represent a complete listing of all reported and potential drug interactions.Drugs that Decrease Calcineurin Inhibitor Concentration by Induction of P-450 Activity. Antituberculous Drugs. Rifampin (and
rifabutin to a lesser extent) markedly reduces cyclosporine and tacrolimus levels, and it may be difficult to achieve therapeutic levels for patients taking rifampin, the use of which should be avoided if at all possible. Pyrazinamide and ethambutol may reduce drug levels, and their use requires monitoring. Isoniazid (INH) can be used with careful drug-level monitoring and is the preferred drug for tuberculosis prophylaxis if this proves essential (see
Chapter 11).
Anticonvulsants. Of the so-called first-generation antiepileptic drugs,
barbiturates markedly reduce cyclosporine and tacrolimus levels. Dose requirements may double or triple, and thrice-daily administration may be required under careful supervision.
Phenytoin and primidone reduce levels and should be used with great care. The average requirement for cyclosporine or tacrolimus is about doubled for patients receiving phenytoin.
Carbamazepine may also decrease cyclosporine levels, but the effect is less pronounced. Benzodiazepines and valproic acid do not affect drug levels, but the latter drug has been associated with hepatotoxicity. Modafinil can cause an up to 50% reduction in calcineurin inhibitor levels. Patients taking these anticonvulsants before transplantation should have a neurologic assessment with a view toward discontinuing them
when possible or exchanging them for one of the new generation of anticonvulsants that do not interact with calcineurin inhibitors.
Of the second generation antiepileptic drugs, oxcarbazepine (Trileptal) may decrease cyclosporine levels. Gabapentin (Neurontin) and levetiracetam (Keppra) and other drugs in this category do not appear to have significant interactions.
Other Drugs. There are isolated reports of several antibiotics, including nafcillin, intravenous trimethoprim, intravenous sulfadimidine, imipenem, cephalosporins, and terbinafine, reducing cyclosporine level. An increased incidence of acute rejection episodes has been described after the introduction of ciprofloxacin. The antidepressant herbal preparation Hypericum perforatum (St. John’s wort) may reduce cyclosporine levels by enzyme induction. Ticlopidine may reduce cyclosporine levels. Cholestyramine, GoLYTELY, sevelamer (Renagel), and olestra may reduce levels by impairing GI absorption. Corticosteroids are inducers of P-450, an effect that needs to be considered if their administration is discontinued. Following cessation of concomitant corticosteroid therapy, tacrolimus levels may increase by up to 25%. The serum creatinine level may increase as a result and lead to a confusing clinical picture.
Prolonged Use. If prolonged use of a drug that induces P-450 activity is required, addition of a drug that inhibits or competes with the P-450 system (e.g., diltiazem, ketoconazole) may facilitate the achievement of therapeutic calcineurin inhibitor levels. Administration of the calcineurin inhibitor on a thrice-daily basis rather than the usual twice-daily basis may also be effective.
Drugs that Increase Calcineurin Inhibitor Levels by Inhibition of P-450 or by Competition for Its Pathways. Calcium Channel Blockers. Verapamil, diltiazem, amlodipine, and nicardipine may significantly increase calcineurin inhibitor levels. Diltiazem and verapamil are sometimes added routinely as adjuncts to the immunosuppressive regimen. Their use may safely permit up to a 40% reduction in the cyclosporine dose. Careful monitoring of drug levels is required when these calcium channel blockers are used for the management of hypertension or heart disease, and physicians and their patients should be specifically warned that changing the dosage of these drugs is equivalent to changing the dosage of the calcineurin inhibitor. Brand-name and generic forms of these drugs (e.g., Cardizem, Dilacor, Tiazac, and Cartia are all forms of diltiazem) may have different effects on calcineurin inhibitor levels. Nifedipine, isradipine, and felodipine have similar hemodynamic effects but have minimal effects on drug levels.
Antifungal Agents. Ketoconazole, fluconazole, itraconazole, and voriconazole markedly elevate calcineurin inhibitor levels. The interaction with ketoconazole is a particularly potent one, which may permit a safe reduction of up to 80% in the cyclosporine or tacrolimus dose. Great care must be taken when stopping and starting these antifungal agents. An important interaction between ketoconazole and histamine blockers has also been described. The effective reabsorption of ketoconazole from the GI tract requires acidic gastric contents, and the addition of a histamine-2 receptor antagonist may reduce its absorption, indirectly producing a clinically significant fall in calcineurin inhibitor levels.
Antibiotics. Erythromycin, even in low doses, may increase calcineurin inhibitor levels. Other macrolide antibiotics (e.g., clarithromycin, josamycin, ponsinomycin) may also increase levels. There are conflicting reports on the impact of
azithromycin on drug levels; however, this drug can generally be given in short courses without monitoring. Because erythromycin is prescribed so ubiquitously, physicians, dentists, and patients should be warned about this interaction. Chloramphenicol may increase tacrolimus levels.
Antiretroviral Therapy. With the advent of highly active antiretroviral therapy (HAART), selected HIV-positive patients may be deemed candidates for kidney transplantation (see
Chapters 7 and
11). Some of the antiretroviral agents, particularly protease inhibitors, are potent inhibitors of P-450.
Ritonavir is the most potent inhibitor of P-450 that is clinically available, and when used alone or in combination (kaletra-retonavir/lopinavir), very small doses of calcineurin inhibitor (e.g., 1 mg/week of tacrolimus) may maintain adequate drug levels. Tenofovir (a component of Truvada and Atrypla) is nephrotoxic and should be avoided after transplantation. Because of multiple drug-drug interactions, immunosuppressive management of HIV-positive patients requires a close and ongoing collaboration and coordination between infectious disease consultants and the transplantation team.
Histamine Blockers. There are conflicting reports regarding the use of cimeti-dine, ranitidine, and omeprazole with calcineurin inhibitors. These drugs may increase creatinine levels without reducing the glomerular filtration rate (GFR) by suppressing proximal tubular creatinine secretion. There may be increased hepatotoxicity when ranitidine and cyclosporine are used in combination.
Hormones. Corticosteroids in high and low doses may decrease the clearance of cyclosporine metabolites. This effect may be particularly pronounced during “pulse” steroid therapy and may result in a confusing clinical picture if the drug levels are measured by a nonspecific assay. Oral contraceptives, anabolic steroids, testosterone, norethisterone, danazol, and somatostatin may also increase drug levels.
Other Drugs. Amiodarone, carvedilol, allopurinol, bromocriptine, and chloro-quine are reported to increase cyclosporine levels. Metoclopramide and grapefruit juice increase the absorption of calcineurin inhibitors (see
Chapter 18).
Drugs that May Exaggerate Calcineurin Inhibitor Nephrotoxicity. Any potentially nephrotoxic drug should be used with caution in combination with the calcineurin inhibitors because the vasoconstrictive effect of the drug tends to potentiate other nephrotoxic mechanisms. Well-substantiated enhanced renal impairment has been described after the introduction of amphotericin and aminoglycosides, and renal impairment may occur earlier than anticipated. Nonsteroidal antiinflammatory drugs should be avoided if possible but can be given for short periods under supervision. Calcineurin inhibitors may potentiate the hemodynamic renal dysfunction seen with angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists. Metoclopramide may increase calcineurin inhibitor levels by increasing its intestinal reabsorption. A syndrome of diarrhea, hepatopathy, and renal dysfunction has been ascribed to the interaction between cyclosporine and colchicine, particularly when given to patients with familial Mediterranean fever.
Lipid-Lowering Agents. The β-hydroxy-β-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (HCRIs) are frequent accompaniments of the immunosuppressive protocol (see
Part IV).
Lovastatin has been implicated in several cases of acute renal failure. When used in full doses in combination with cyclosporine, lovastatin can cause rhabdomyolysis with elevated creatine phosphokinase
levels and acute renal failure. Myopathy alone has been observed in up to 30% of recipients of the lovastatin-cyclosporine combination, with symptoms of muscle pain and tenderness developing 6 weeks to 16 months after commencement of therapy. The myopathic syndrome has not been observed when lova-statin is used in a daily dose of 20 mg or less. Even this dose should be used with caution, however, and patients should be made aware of the potential interaction. The coadministration of lovastatin with gemfibrozil further increases the likelihood of rhabdomyolysis. The newer HCRIs—pravastatin, fluvastatin, simvastatin, atorvastatin, rosuvastatin—should be introduced at low doses and maximal doses avoided. Cyclosporine may increase the levels of eze-timibe, but ezetimibe has not been reported to affect the levels of cyclosporine. Cholestyramine may interfere with cyclosporine absorption from the GI tract.
Side Effects
Nephrotoxicity. Nephrotoxicity is the major “thorn in the side” of these remarkable drugs. Theories linking the mechanism of immunosuppression and nephrotoxicity are discussed later. The terms
cyclosporine and
FK toxicity are often used loosely, and it is important to note that these terms encompass several distinct, overlapping syndromes (
Table 5.2). Readers are referred to the extensive review of this topic by Naesens and colleagues (see “
Selected Readings”).
Functional Decrease in Renal Blood Flow and Filtration Rate. The calcineurin inhibitors produce a dose-related, reversible, renal vasoconstriction that particularly affects the afferent arteriole (
Fig. 5.2). The glomerular capillary ultrafiltration coefficient (Kf) also decreases, possibly as a result of increased mesangial cell contractility. Most of the studies on the mechanism of this effect have used cyclosporine rather than tacrolimus. The picture is reminiscent of “prerenal” dysfunction, and in the acute phase, tubular function is intact.
The normal regulation of the glomerular microcirculation depends on a complex, hormonally mediated balance between vasoconstriction and vasodilation. Cyclosporine-induced vasoconstriction is caused, at least in part, by alteration of arachidonic acid metabolism in favor of the vasoconstrictor thromboxane. Cyclosporine is also a potential inducer of the powerful vasoconstrictor endothelin, and circulating endothelin levels are elevated in its presence. Cyclosporine-induced changes in glomerular hemodynamics can be reversed by specific endothelin inhibitors and by antiendothelin antibodies. The sympathetic nervous system is also activated.
Several in vivo and in vitro studies have suggested that alterations in the L-arginine nitric oxide (NO) pathway may be involved in calcineurin-induced renal vasoconstriction. NO causes relaxation of preglomerular arteries and improves renal blood flow. The constitutive enzyme endothelial nitric oxide synthase (NOS) is produced by renal endothelial cells and modulates vascular tone. Both acute and chronic cyclosporine toxicity can be enhanced by NOS inhibition with N-nitro-L-arginine-methyl ester and ameliorated by supplementation with L-arginine. Interestingly, sildenafil (Viagra) increases GFR in transplant patients, presumably by reversing this effect.
Calcineurin inhibitor-induced renal vasoconstriction may manifest clinically as delayed recovery of early malfunctioning grafts or as a transient, reversible, dose-dependent, blood-level-dependent elevation in serum creatinine concentration that may be difficult to distinguish from other causes of graft dysfunction. Vasoconstriction may be a reversible component of chronic calcineurin inhibitor toxicity, which may amplify the functional severity of the chronic histologic changes seen with prolonged use. The vasoconstriction may be more pronounced with cyclosporine than with tacrolimus and also helps to account for the hypertension and the tendency for sodium retention that are commonly associated with cyclosporine use.
Chronic Interstitial Fibrosis. Interstitial fibrosis, which may be patchy or “striped” and associated with arteriolar lesions (see
Chapter 14), is a common feature of long-term calcineurin inhibitor use. This lesion may produce chronic renal failure in recipients of organ transplants; however, several long-term studies show that in the dose regimens currently employed, kidney function may remain stable, although often impaired, for many years. The mechanism of calcineurin inhibitor-induced interstitial fibrosis remains poorly defined.
Evidence from experimental models suggests that chronic nephropathy involves an angiotensin-dependent up-regulation of molecules that are important in the scarring process, such as TGF-β and osteopontin. Enhanced production of TGF-β in normal T cells may provide the link between the immunosuppressive effects of the calcineurin inhibitors and their nephrotoxicity, and variation in fibrogenic gene expression may help explain the varying consistency of this
effect. Calcineurin-inhibitor induced hypomagnesemia may induce interstitial inflammation and enhance the production of TGF-β, thereby perpetuating chronic fibrotic lesions. Interstitial fibrosis may also be a reflection of intense and prolonged vasoconstriction of the renal microcirculation. Cyclosporine may also impair the regenerative capacity of microvascular endothelial cells and induce apoptosis. The resulting chronic renal ischemia may enhance the synthesis and accumulation of extracellular matrix proteins in the interstitium.
Acute Microvascular Disease. Thrombotic microangiopathy (TMA) (see
Chapters 9 and
14) is a distinct form of calcineurin inhibitor vascular toxicity that may manifest as renal involvement alone or as a systemic illness. It produces a syndrome reminiscent of thrombotic thrombocytopenic purpura (TTP). In TTP, potentially pathogenic inhibitory antibodies against the von Willebrand factor (vWF)-cleaving protease ADAMTS13, a zinc metalloprotease, have been detected. A similar mechanism has been described in calcineurin inhibitor-induced TMA.
Electrolyte Abnormalities and Hypertension. Impaired sodium excretion is a reflection of the renal vasoconstrictive effect of the calcineurin inhibitors. Patients receiving long-term cyclosporine therapy tend to be hypertensive (see
Chapter 10) and to retain fluid. Studies show activation of the renin-angiotensin-aldosterone system and sympathetic nervous system and suppression of atrial natriuretic factor, which results in attenuation of the natriuretic and diuretic response to an acute volume load. NO production is also impaired. Hypertension tends to be less marked (or the need for antihypertensive drugs may be less) for patients receiving tacrolimus, possibly because it produces less peripheral vasoconstriction than does cyclosporine.
Hyperkalemia is common and occasionally requires treatment, although it is rarely life-threatening as long as kidney function remains good. It is not uncommon for patients taking calcineurin inhibitors to have potassium levels in the mid-fives. Hyperkalemia is often associated with a mild
hyperchloremic acidosis and an intact capacity to excrete acid urine. The clinical picture is thus reminiscent of type IV renal tubular acidosis. Patients receiving cyclosporine may have an impaired capacity to excrete an acute potassium load, and there is evidence to suggest impaired production of aldosterone, an acquired impaired renal response to its action, and inhibition of cortical collecting duct potassium secretory channels. Hyperkalemia may be exaggerated by concomitant administration of β blockers, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers. A defect of collecting tubule hydrogen ion secretion has been described with tacrolimus. Both drugs are magnesuric and hypercalciuric, and hypomagnesemia is commonly associated with their use. In liver transplantation, hypomagnesemia may predispose patients to seizures; this has been observed rarely in kidney recipients. The urinary loss of Ca
2+ and Mg
2+ is due to down-regulation of specific transport proteins. Magnesium supplements are often prescribed but may be ineffective because of a lowered renal magnesium threshold (see
Chapter 19). Both cyclosporine and tacrolimus can produce hyperuricemia, although only cyclosporine has been associated with gout, which may resolve when cyclosporine is switched to tacrolimus.
Methods of Amelioration. The vexing issue of calcineurin inhibitor nephrotoxicity has spawned a variety of clinical and experimental approaches designed to modify the renal effects of these drugs, particularly their capacity to produce vasoconstriction. Low-dose dopamine is used in some centers in the early postoperative period to “encourage” urine output. Calcium channel blockers given to both the donor (see
Chapter 4) and the recipient (see
Part IV) may reduce
the incidence and severity of delayed graft function. Omega-3 fatty acids in the form of 6 g of fish oil each day were thought to increase renal blood flow and GFR by reversing the cyclosporine-induced imbalance between the synthesis of vasodilator and vasoconstrictor prostaglandins, but long-term studies have shown no such benefit. The prostaglandin agonist misoprostol and thromboxane synthetase inhibitors may have a similar effect. Various protocol adjustments, discussed later in this chapter, can also be employed to minimize calcineurin inhibitor toxicity.
Nonrenal Calcineurin Inhibitor Toxicity. Gastrointestinal. Episodes of hepatic dysfunction typically manifesting as subclinical, mild, self-limited, dose-dependent elevations of serum aminotransferase levels with mild hyperbilirubinemia may occur in nearly half of all kidney transplant recipients taking cyclosporine and occur less frequently in those taking tacrolimus. No specific hepatic histologic lesion has been described in humans, and the hyperbilirubinemia is a reflection of disturbed bile secretion rather than hepatocellular damage. Cyclosporine does not itself produce progressive liver disease; other causes, most frequently one of the viral hepatitides, need to be considered when this occurs. Cyclosporine therapy is associated with an increased incidence of cholelithiasis, presumably resulting from an increased lithogenicity of cyclosporine-containing bile. Varying degrees of anorexia, nausea, vomiting, diarrhea, and abdominal discomfort occur in up to 75% of patients receiving tacrolimus, and less frequently in patients receiving cyclosporine.
Cosmetic. The cosmetic complications of cyclosporine must be treated seriously, particularly in women and adolescents, because of the misery they can produce and the temptation to resolve them through noncompliant behavior. Cosmetic complications are often exaggerated by concomitant use of corticosteroids. They are less prominent for patients receiving tacrolimus.
Hypertrichosis in varying degrees occurs in nearly all patients receiving cyclosporine and is particularly obvious in dark-haired girls and women. A coarsening of facial features is observed in children and young adults, with thickening of the skin and prominence of the brow. Tacrolimus may produce hair loss and frank alopecia. Gingival hyperplasia, which can be severe, may develop in patients receiving cyclosporine and is exaggerated by poor dental hygiene and possibly by concomitant use of calcium channel blockers. Azithromycin, a macrolide antibiotic that does not affect cyclosporine metabolism, may reduce gingival hyperplasia. Gingivectomy may occasionally be indicated, and switching from cyclosporine to tacrolimus is usually effective. Cosmetic complications tend to become less prominent with time. Sympathetic cosmetic counseling is required. Cyclosporine may increase prolactin levels, occasionally producing gynecomastia in men and breast enlargement in women.
Hyperlipidemia. Cyclosporine has been implicated as one of the various factors responsible for the generation of post-transplantation hypercholesterolemia (see
Chapter 10). The mechanism of this effect may be related to abnormal low-density lipoprotein feedback control by the liver, to altered bile acid synthesis, or to occupation of the low density lipoprotein receptor by cyclosporine. Up to two thirds of patients develop
de novo hyperlipidemia in the first posttransplantation year. The effect is less marked with tacrolimus, and lipid levels may decrease when patients are switched from cyclosporine to tacrolimus.
Glucose Intolerance. Post-transplantation glucose intolerance and new-onset diabetes mellitus (NODM) are discussed in
Chapter 10. Both calcineurin inhibitors
are toxic to pancreatic islets, although tacrolimus is more so, possibly as a result of increased concentrations in islets of FKBP relative to cyclophilin. The effect is dose related and may be exaggerated by concomitant corticosteroid use. Morphologic changes in the islets include cytoplasmic swelling, vacuolization, and apoptosis, with abnormal immunostaining for insulin. Obesity, African American or Hispanic ethnicity, family history of diabetes, and hepatitis C infection may predispose to NODM.
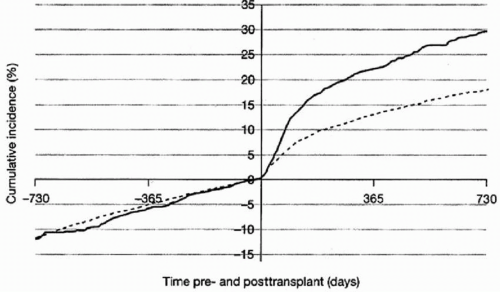
Figure 5.3 shows the incidence of diabetes before and after transplantation by type of calcineurin inhibitor as reported to the United States Renal Data System.
Neurotoxicity. A spectrum of neurologic complications has been observed in patients receiving calcineurin inhibitors; they are generally more marked with tacrolimus. Coarse tremor, dysesthesias, headache, and insomnia are common and may be dose and blood-level related. Patients may complain of discrete cognitive difficulties coinciding with peak drug levels. More severe complications are uncommon in kidney recipients, although isolated seizures may occasionally occur, and full-blown leukoencephalopathy has been described. Bone pain in long bones has also been described.
Infection and Malignancy. Infection and malignancy inevitably accompany immunosuppression and are discussed in detail in
Chapters 10 and
11. Despite their immunosuppressive potency, the incidence of infections and common
de novo neoplasms has not significantly increased since the introduction of the calcineurin inhibitors, although the course of malignancies may be accelerated.
Thromboembolism. In vitro, cyclosporine increases adenosine diphosphate-induced platelet aggregation, thromboplastin generation, and factor VII activity. It also reduces production of endothelial prostacyclin. These findings may be causally related to the somewhat increased incidence of thromboembolic events
that have been observed in cyclosporine-treated kidney transplant recipients. The finding of glomerular microthrombi as part of calcineurin inhibitor-induced microangiopathy was discussed previously.
Hyperuricemia and Gout. Hyperuricemia, because of reduced renal uric acid clearance, is a common complication of calcineurin inhibitor therapy, particularly when diuretics are also employed. Episodes of gout are more common in patients receiving cyclosporine than tacrolimus and have been reported in up to 7% of patients. Treatment is discussed in
Chapter 10.