Features
Classical AID
PSC
Autoantigen(s)
Yes
Possibly
Autoantibody
Yes, pathogenetic
Yes, biomarker
Age
Children and adults
Children and adults
Gender predilection
Female > male
Male > female
Genetic factors
HLA, non-HLA
HLA, non-HLA
Tissue- or organ-specific disease
Yes
Yes
Inflammatory cells
Autoreactive T cells
Gut-primed T cells, NK, NKT, macrophages, γδ T cells
Environmental factors
Yes
Yes
Associated AIDs
Yes
Yes
Response to immunosuppression
Yes
No
Examples
SLE
Myasthenia gravis
Graves’ disease
Pernicious anemia
Type 1 diabetes
AIH
PBC
A form of secondary sclerosing cholangitis associated with elevations of serum IgG4 and/or IgG4-secreting B and plasma cells may mimic PSC [5]. Retrospective studies indicate that approximately 10 % of patients diagnosed with PSC instead may have IgG4 cholangiopathy [6]. IgG4 cholangiopathy can be distinguished by a prior history of pancreatitis, stricturing of both intrahepatic and extrahepatic bile ducts, propensity for jaundice, and the use of recently developed techniques [7].
Multiple immunological features suggest involvement of innate and adaptive immune responses in immunopathogenesis, including susceptibility and resistance associations with HLA haplotypes and autoantibodies (autoAbs), and evidence that gut-primed T effector T cells mediate peribiliary, fibrosing inflammation [4, 8]. The homing and retention of these gut-primed T cells are facilitated by the activated cholangiocytes that express ligands and receptors and secretion of inflammatory cytokines and chemokines [9]. Thus, the cholangiocytes are not passive targets of the immune response but participate in the immunopathogenesis of PSC [4].
The goal of this chapter is to provide a progress report on the immunology of PSC. Emphasis is placed on immunological findings advancing our understanding of the immunopathogenesis of PSC.
Biliary Anatomic Features and PSC
The branching network of bile ducts is lined by cholangiocytes with tight junctions that retain bile within the duct lumens (Fig. 9.1) [10, 11]. Each bile duct is accompanied by a branch of the hepatic artery of equal caliber that gives rise to a peribiliary capillary plexus surrounding each duct. Lymphatic channels adjacent to the peribiliary capillaries drain lymph formed in the space of Disse that contains cytokines and other constituents produced in the hepatic lobules. The portal venous blood from the small bowel and colon contains pathogen-associated molecular patterns (PAMPs) from the cell walls and unmethylated DNA of gut bacteria and fungi, metabolites produced by the gut microbiota, and viable microbial pathogens when the gut mucosal barrier is breached. PSC markedly alters these homeostatic anatomic relationships.

Fig. 9.1
Biliary anatomic features involved in primary sclerosing cholangitis. An intralobular bile duct receives the bile secreted by hepatocytes through cholangioles at the periphery of the portal tract. Each intrahepatic bile duct is accompanied by a branch of the hepatic artery of equal caliber. The arteries supply a peribiliary capillary plexus surrounding each duct, while lymphatic channels lie adjacent to the peribiliary capillaries and drain lymph formed in the space of Disse that contains cytokines and other constituents produced in the hepatic lobules. The portal venous blood from the small bowel and colon contains pathogen-associated molecular patterns (PAMPs) from the cell walls and unmethylated DNA of gut bacteria and fungi, metabolites produced by the gut microbiota, and viable microbial pathogens when the gut mucosal barrier is breached
Pathology of PSC
The histopathology of PSC is unique among primary biliary tract diseases (Fig. 9.2) [12]. Lymphoplasmacytic infiltrates of the portal tracts localize to the peribiliary space, where they promote peribiliary fibrosis without apoptotic destruction of the cholangiocytes. The density of portal inflammation is scant, especially when compared to either autoimmune hepatitis (AIH) or primary biliary cholangitis (PBC). A key feature distinguishing PSC from PBC is the absence of effector cell-mediated apoptosis of cholangiocytes in PSC [13].

Fig. 9.2
Histopathology of small duct and medium duct primary sclerosing cholangitis. The histopathology of PSC includes the small duct variant and the fibrous inflammatory lesions of medium-caliber intrahepatic ducts. Compared to either AIH or PBC, the inflammatory infiltrates in PBC are sparse. Periductal, concentric fibrosis of the medium-caliber intralobular bile ducts pushes the peribiliary capillary plexi away from the basement membranes of the bile ducts
Progressive fibrosis leads to concentric, circumferential laminations around intact intrahepatic bile ducts, referred to as “onion skin” fibrosis, that displace the peribiliary capillary plexi, creating a physical and spatial barrier to oxygenation and maintenance of the cholehepatic countercurrent circulation between the bile duct and artery [14]. Thus, the pathogenesis of stricturing, circumferential peribiliary fibrosis also involves relative arterial or capillary ischemia. Stimuli of periductal fibrosis include secretion of chemokines and cytokines by innate immune cells and activated cholangiocytes and the inflammatory and fibrotic response to toxic bile leaking between injured cholangiocytes [4, 15]. Proinflammatory cytokines and/or microbial molecules in lymph or blood induce cholangiocyte expression of chemokine receptors and secretion of chemokines and cytokines involved in the chemoattraction of effector cells to the peribiliary space and promotion of fibrogenesis [4, 16].
Innate and Adaptive Immunity
Innate Immunity
Innate immunity provides immediate reactions against microbial pathogens and cells altered by stress, infection, or neoplasia [17, 18]. Innate immune responses are mediated by macrophages (including Kupffer cells), dendritic cells (DCs), natural killer (NK), and NKT cells. Macrophages and DCs constitutively express pattern recognition receptors (PRRs) for invariant microbial molecules, collectively called PAMPs, and for CD14 and activated complement (C’) molecules. Toll-like receptors (TLRs) are the most prominent PRRs, expressed on innate immune cells and epithelial cells, including cholangiocytes and hepatocytes. Since PAMPs are molecular fragments of microbes, innate immune responses do not require viable microbes. PAMPs relevant to the immunopathogenesis of PSC [11, 19] include (1) lipopolysaccharide (LPS, aka endotoxin), the signature cell wall component of all Gram-negative bacteria; (2) lipoteichoic acid, the signature cell wall component of Gram-positive bacteria; (3) peptidoglycans, essential cell wall components of all bacteria; and (4) unmethylated, bacterial CpG dinucleotide motifs. Class I chain-related MICA and MICB genes encode ligands expressed by cells damaged by stress, infection, or neoplasia that bind to NKG2D receptors on NK cells, NKT cells, macrophages, and γδT cells causing target cell lysis. In addition, MICA ligands also costimulate CD8 CTLs through their NKG2D receptors.
Innate Immunity in PSC
Intense, unregulated innate immune responses are involved in PSC immunopathogenesis [4, 20]. The cholangiocytes of PSC patients express normal amounts of TLR4 and nucleotide-binding oligomerization domain-containing protein (NOD)-like receptor family pyrin domain-containing 3 (NLRP3) but excessive TLR9 [21]. TLR9 expression correlated with fibrosis stages and greater risk for orthotopic liver transplantation (OLT). Cholangiocytes activated by TLRs, proinflammatory cytokines, and interferon-γ (IFNγ) produce cytokines and chemokines involved in the peribiliary localization of specific inflammatory cells and peribiliary fibrogenesis (Fig. 9.3, discussed below) [4, 9, 22].
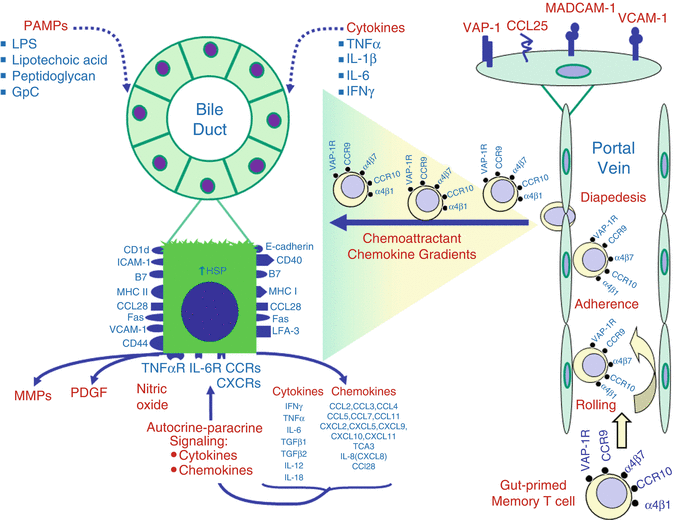
Fig. 9.3
Activated cholangiocytes and gut-primed T cells in the immunopathogenesis of primary sclerosing cholangitis. Cholangiocytes are activated by PAMPs and by proinflammatory cytokines TNFα, IL-1β, IL-6, and IFNγ. Activated gene expression leads to cholangiocyte production of multiple immunological ligands and receptors, chemokines, cytokines, MMPs, PDGF, NO, and aberrant class II HLA. In PSC, cholangiocytes secrete the chemokine CCL25, the ligand for CCR9 on gut-primed T cells. Portal endothelial cells in PSC livers express VAP-1, whose amine oxidase function in the presence of proinflammatory cytokines, especially TNFα, results in aberrant expression of MADCAM-1 and display of CCL25. This permits adhesion and diapedesis of gut-primed memory T cells bearing the α1β7 integrin receptor for MADCAM-1 and the chemokine receptor CCR9 for the CCL25. After transendothelial migration, gut-primed memory T cells migrate along the gradients of chemokines secreted by activated cholangiocytes to congregate in the peribiliary space. The chemokine CCL28 facilitates peribiliary recruitment of T cells bearing its chemokine receptor, CCR10, while VCAM-1 on the cell surface of cholangiocytes acts as ligand for the T cell integrin receptor α1β4. This postulated scheme does not require the presence of gut Ag(s) that originally primed the T cells in the GALT. The absence of cholangiocyte expression of the priming gut Ag(s) may explain the observation that peribiliary T cells do not cause apoptosis of cholangiocytes in PSC
Adaptive Immunity
Adaptive immunity involves delayed immune responses of T cell receptors (TCRs) to processed peptide antigens (Ags, potentially including autoAgs) presented within Ag-binding grooves of class I and II major histocompatibility complex molecules (MHC, designated HLA in humans) expressed by professional antigen-presenting cells (APCs) [23, 24]. Professional APCs include DCs of the innate immune system and activated B cells. CD4 T cell TCRs react with processed exogenous Ags presented by class II MHC molecules and stimulate Ag-specific CD4 T cell TCRs, while CD8 TCRs are stimulated by endogenous (including viral) Ags presented by class I MHC molecules. MHC binding of specific peptide Ags is genetically determined [25, 26]. The non-polymorphic MHC class I-like molecule, CD-1, presents lipid Ags to TCRs expressed by γδT cells. γδT cells are involved in mucosal immunity, surveillance of neoplastic changes, and protection from autoimmune diseases and microbial infections [27]. Class III MHC genes encode TNFα/β; C’ factors C4, C2, and Bf; as well as heat shock proteins [25, 26].
HLA
HLA genes are inherited from each parent to form haplotype pairs [25, 26]. Class I HLA, expressed by HLA-A, HLA-B, and HLA-Cw loci, presents peptide Ags to TCRs of cytotoxic CD8 T cells. Class II HLA, expressed by HLA-DR, HLA-DQ, and HLA-DP loci, presents processed peptide Ags to TCRs of CD4 T cells. Polymorphic HLA class I and II Ag-binding grooves determine whether binding and presentation of specific peptide Ags occur, thus conferring susceptibility or resistance to development of a disease like PSC. The class III locus encodes polymorphic immune response proteins, including TNFα/β, complement (C’) factors, and heat shock proteins.
Effector T Cells and Cytokines
Ag activation of CD4 T helper (Th) cells triggers exclusive pathways of differentiation that generate Ag-specific Th1, Th2, Th17, Th9, and T follicular helper (Tfh) cells and T regulatory (Treg) subsets [28]. A milieu containing IL-12, IL-18, and INFγ favors CD4 differentiation into Th1 cells that secrete the signature cytokines of Th1 cells: IL-2, INFγ, and TNFα/β. Th1 cytokines provide help for proliferation and differentiation of CD8 T cells, also called cytotoxic T lymphocytes (CTLs), and activate macrophages. Th1 cytokines also induce B cell secretion of C’-fixing IgG2a. In contrast, a milieu containing IL-4 favors CD4 differentiation into Th2 that secretes the signature cytokine profile of Th2 cells, IL, and activates eosinophils and mast cells. The signature cytokines of CD4 Th1 inhibit the proliferation of Th2 cells and vice versa, creating a dynamic balance between Th1 and Th2 subsets within inflammatory infiltrates. Transforming growth factor-beta (TGFβ), IL-6, IL-21, IL-23, and retinoic acid receptor-related orphan receptors γ and α (RORγ, RORα) promote generation of Th17 cells that can become either protective or pathogenic. Both Th1 IFNγ and Th2 IL-4 inhibit Th17 differentiation. Pathogenic Th17 cells are induced by IL-23 and IL-1β to secrete IL-17A, IL-17F, IL-21, and IL-22. In autoimmunity, Th17 effector cells intensify and perpetuate tissue inflammation. Th9 cells have not been evaluated in PSC; however, several functions indicate that they may be relevant to immunopathogenesis [29]. For example, secretion of IL-9 increases gut permeability, activates mast cells, and increases leukocyte recruitment. Th9 cells also secrete IL-21, which promotes IFNγ production by NK cells and CD8 T cells, and IL-3, which enhances DC survival. Tfh cells localize within B cell follicles in lymph nodes and Peyer’s patches, where they promote selection and survival of B cell clones by expression of CD40 ligand and secretion of IL-4 and IL-21 [30].
CD4 Treg cells mediate Ag-specific suppression of T cell responses by local secretion of IL-10 and transforming growth factor-beta (TGFβ) [28]. The protective Th17 subset of Treg17 cells is induced by IL-6 and TGFβ.
Adaptive Immunity in PSC
Recent studies have focused on the role and functions of Tregs in PSC. Genome-wide association studies (GWAS) identified single nucleotide polymorphisms (SNPs) that could affect Treg cells, which led to studies of circulating and hepatic quantities of CD4+-CD25high-FOXP3+-CD127low Tregs [31]. Tregs were significantly decreased in the blood and liver, and their suppressor function was reduced. Reduced Tregs in the blood significantly correlated with homozygosity for the major allele of the SNP rs10905718 in the IL-2RA gene. These findings provide a genetic basis for immune dysregulation caused by reduced Treg numbers in PSC. Another study of Tregs in peripheral blood mononuclear cells (PBMC) of patients with concurrent PSC and UC showed higher frequencies of Tregs compared to those in patients with UC alone [32].
Among the autoAbs associated with PSC is an IgA anti-cholangiocyte Ab, which occurs at high frequency and is correlated with more rapid progression to death or OLT compared to PSC patients without this autoAb [33]. The signature cytokine of Th17 cells, IL-17A, promotes hepatic inflammation and fibrosis [34]. To investigate Th17 immune responses to pathogens in PSC, hepatic bile obtained using endoscopic retrograde cholangiopancreatography (ERCP) was cultured, and liver biopsies were stained using 16sRNA fluorescence in situ hybridization (FISH) [34]. The bile grew multiple bacterial and fungal species and FISH detected microbes in 12 of 13 (92 %) of portal tracts. Stimulation PBMC with microbes cultured from the bile generated high frequencies of Th17 cells, especially in response to Candida albicans. Th17 cells expressing IL-17A were detected in the peribiliary space, indicating a pathogenic role in the generation of fibrosing inflammation.
Transendothelial Leukocyte Trafficking into Tissues
Activated, circulating leukocytes enter tissues by a multistep process of transendothelial migration [8, 9, 16]. Cellular injury or stress causes secretion of chemokines that are taken up by endothelial cells and displayed on their luminal surfaces along with adhesion molecules. As circulating, activated leukocytes expressing chemokine receptors and counter-receptors for adhesion molecules encounter activated endothelial cells, their leukocyte selectin receptors cause them to roll along the endothelium. Rolling ceases when firm leukocyte adhesion occurs due to binding of leukocyte chemokine receptors to chemokines displayed by endothelial cells and leukocyte integrin adhesion molecules to endothelial cellular adhesion molecules. This initiates diapedesis of leukocytes through endothelial tight junctions and basement membranes into the tissue, where they are chemoattracted along the chemokine gradient toward the source of chemokine secretion. Thus, both chemokines and adhesion molecules expressed on the endothelium determine the composition of inflammatory infiltrates entering the tissue from the blood. As discussed below, this process appears to play a key role in the immunopathogenesis of PSC [8, 9, 16].
Progress Toward an Understanding of Immunopathogenesis
Genetics
Genome-Wide Association Studies (GWAS)
Genetic susceptibility to PSC was assessed in a GWAS of 443,816 single nucleotide polymorphisms (SNPs) in 285 Norwegian PSC patients and 298 healthy controls [35]. Detected associations were reassessed in independent case-control panels in 766 PSC patients and 2,935 controls from Scandinavia, Belgium, the Netherlands, and Germany. The strongest associations were near the HLA-B locus (rs3099844, OR −4.8, 95 % CI 3.6–6.5, p = 2.6 × 10−26, and rs2844559, OR 4.7, 95 % CI 3.5–6.4, p = 4.2 × 10−26). Non-HLA rs9524260 on chromosome 13q31 was significantly associated with three of four groups. This locus encodes glycan 6, and inhibition of glycan 6 in a cholangiocyte cell line resulted in upregulation of proinflammatory markers.
Subsequent dense genotyping of 130,422 SNPs in immune-related disease regions was performed in 3,789 PSC patients of European ancestry and compared with 2,079 controls [36]. In addition to confirming three significant non-HLA associations, nine new non-HLA associations were detected. Six of the nine were more strongly associated with PSC than with comorbid IBD. These studies have expanded the genetic risk map of PSC, providing a better understanding of the relationship of PSC and other immune-mediated diseases.
Fucosyltransferase 2 (FUT2)
FUT2 introduces fucose into glycoproteins and glycolipids. FUT2 activity influences interactions between the host and microbes [37]. The nonsense mutation G428A and missense mutation A385T are the principal variants that cause 20 % of people to be FUT2 “nonsecretors,” incapable of secreting fucose-containing Ags and lacking epithelial cell fucosylation. GWAS indicated that inactivating FUT2 variants were associated with PSC, Crohn’s disease, and biochemical markers of biliary injury [37]. The microbiome of nonsecretors was characterized by reduced bifidobacteria, increased Firmicutes, and decreased Proteobacteria and Actinobacteria. The bacterial content of the bile also differed from that of secretors. Lack of fucosylated glycans on the surface of cholangiocytes is potentially deleterious because it would interrupt the glycocalyx required for the protective biliary bicarbonate umbrella that shields cholangiocytes from hydrophobic bile salt toxicity.
HLA and Susceptibility to PSC
PSC susceptibility is most strongly associated with four distinct HLA haplotypes (Table 9.2) [35, 38–41]. The highest susceptibility is conferred by homozygosity for MICA*008 (OR 5.01), suggesting that this allele is closely linked to a true susceptibility allele [42]. The MICA*008 allele contains the MICA5.1 microsatellite allele, which explains the microsatellite’s significant association with PSC. It is possible that the NKG2D ligand produced by the MICA*008 allele might explain the increased numbers of NK and γδT cells in PSC livers [43, 44]. The MICB microsatellite allele MICB24 is also significantly associated with PSC. Of note, PSC associations with both MICA5.1 and MICB24 microsatellites are observed exclusively with the HLA-B8-DR3 haplotype [45].
Table 9.2
Immunogenetic associations of PSC with HLA and non-HLA alleles
Susceptibility haplotypes | Odds ratio |
B8-MICA*008-TNFA*2-DRB3*0101-DRB1*0301-DQA1*0501 DQB1*0201 | 2.69 |
DRB3*0101-DRB1*1301-DQA1*0103-DQB1*0603 | 3.80 |
MICA*008-DRB5*0101-DRB1*1501-DQA1*0102-DQB1*0602 | 1.52 |
(MICA*008 homozygosity) | 5.01 |
Resistance haplotypes | |
DRB4*-DRB1*0401-DQA1*0301-DQB1*0302 | 0.26 |
DRB4*-DRB1*0701-DQA1*0201-DQB1*0303 | 0.15 |
MICA*002 | 0.12 |
Non–MHC associations | |
ICAM-1 | NA |
MMP-1, MMP-3 | NA |
CTLA4 | NA |
CCR5Δ32 deletion | NA |
CFTR | NA |
The fact that the HLA-DR3 haplotype is absent from the other two HLA haplotypes associated with the second greatest susceptibility risk (OR 3.80) has been interpreted as evidence of linkage disequilibrium among HLA-B8, MICA*008, TNFα promoter (TNFA*2), and a yet unidentified susceptibility allele. Since DRB1 alleles are present in all three extended susceptibility HLA haplotypes, V or G at position 86 of the DRβ chain was analyzed. V86 was associated with susceptibility alleles DRB1*0301, DRB1*1301, and DRB1*1501 (OR 3.01), while G86 was associated with resistance alleles DRB1*0401 and DRB1*04 (OR 0.17). Modeling of susceptibility and resistance indicated that K87 and P55 in the DQB also could explain susceptibility (OR 2.78) or resistance (OR 0.28).
Of interest, one of the HLA susceptibility haplotypes contains the TNFA*2 allele (Table 9.2). Autoimmunity is associated with TNF-2 allele -308A [46], but a G-308A substitution in the TNFα promoter is linked with susceptibility only with the DRB3*0101 haplotype [47]. PSC susceptibility was not associated with the A to G polymorphism of Fas (encoded by the TNFSF6 gene) [48].
A single HLA susceptibility allele may exist in PSC, but it is more likely that PSC susceptibility is genetically complex, involving multiple HLA and non-HLA SNPs. Currently, PSC susceptibility can be explained for only 50 % of PSC cases on the basis of any allele, amino acid substitutions in the DRβ peptide, or homozygosity for MICA*008 [38]. This is independent of IBD, since UC is unassociated with these HLA haplotypes or MICA*008. Further investigations will require studies of SNPs identified in GWAS.
Susceptibility associations of HLA-DR3 and class III TNFA*2 and the G-308A substitution in the TNFα promoter may explain the association of PSC with AIDs [49]. HLA-DR3+ leukocytes secrete significantly greater amounts of IL-2, IL-5, IL-12, and IFNγ than do HLA-DR3− leukocytes, before and after mitogen stimulation in vitro [50]. In contrast, HLA-DR3 haplotype does not influence secretion of anti-inflammatory Th2 cytokines IL-4 or IL-10. Susceptibility for PSC may reflect overproduction of TNFα and IFNγ. If high levels of these cytokines are obligatory for immunopathogenesis, it would be plausible that patients capable of generating similar levels of cytokines might develop PSC in the absence of HLA-DR3.
Non-MHC Genes and Susceptibility to PSC
Polymorphic non-HLA gene products involved in inflammation and immunoregulation may be biomarkers of progression and severity of PSC. No susceptibility associations have been identified for Nod2, IL-1, IL-1B, and IL-RN [19, 48]. CTLA4, a T cell receptor for costimulatory B7 ligands that downregulates T cell activation, is of great interest, since CTLA4 polymorphisms increase the risk of multiple organ-specific AIDs [51]. Susceptibility for PSC remains controversial, being present in one study and not in another [48]. The mutant chemokine receptor 5 with a deletion of 32 base pairs (CCR5Δ32) has reduced expression and function. Although initial results were controversial, a recent study showed that PSC susceptibility was significantly associated with CCR5Δ32 [52]. Fibrosis results from a dynamic imbalance between matrix metalloproteinases (MMPs) and inhibitors of metalloproteinases. The MMP-3 gene, encoding stromelysin, exhibits a promoter sequence polymorphism (5A or 6A repeat). A 5A allelic association was observed in one study but was not confirmed in another [53, 54]. The 5A allele was found more frequently in PSC patients with UC (60 %) than in PSC alone (45 %) [54]. The MMP-9 polymorphism R279Q was significantly associated with susceptibility [55]. No association was noted with MMP-1 promoter polymorphisms [54]. The TGFB1 gene encoding the profibrotic and immunosuppressive cytokine TGFβ was not associated with PSC [48]. The absence of the murine bile transporter, Mdr2 (Abcb4), caused regurgitation of toxic bile through leaky cholangiocyte tight junctions, resulting in PSC-like lesions. In contrast, PSC is characterized by normal bile acid transporter haplotypes for MDR3 (human homolog of murine Mdr2), ABCB4, and bile salt export protein (BSEP) ABCB11; thus, there is no evidence of a susceptibility association [56]. Of note, claudin-1 gene mutations compromise tight junctions and are associated with neonatal ichthyosis and sclerosing cholangitis [57]. PSC-like lesions in cystic fibrosis prompted testing for mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). One report indicated an increased prevalence of CFTR mutations and defective nasal CFTR Cl− channel function [58], but others failed to confirm these findings [59]. Induction of experimental colitis in cftr−/− knockout mice did cause PSC-like lesions, suggesting that CFTR mutations might contribute to pathogenesis of PSC in the presence of active IBD [60].
MHC Genes and Resistance to PSC
Three HLA haplotypes reduce the risk of PSC (Table 9.2). HLA-DR4 is the most protective; however, when PSC occurs in HLA-DR4-positive patients, they paradoxically have poorer prognosis and an increased risk of cholangiocarcinoma [61]. One copy of either the MICA*002 allele or its satellite allele MICA9 also confers significant resistance [42, 45]. Given the strong susceptibility risk of PSC bestowed by MICA*008, the resistance association with MICA*002 strongly suggests that MICA-encoded ligands for the NKG2D receptors of innate immune-responsive cells and CD8 CTLs are determinants of the immunopathogenesis of PSC. MICA allelic associations also imply involvement of innate immune effector cells and microbial PAMPs in immunopathogenesis.
Related posts:
 Pediatric Primary Sclerosing Cholangitis
Pediatric Primary Sclerosing Cholangitis
 Pruritus in Primary Sclerosing Cholangitis: New Insights into Cause and Treatment
Pruritus in Primary Sclerosing Cholangitis: New Insights into Cause and Treatment
 Ursodeoxycholic Acid Treatment in Primary Sclerosing Cholangitis
Ursodeoxycholic Acid Treatment in Primary Sclerosing Cholangitis
 Percutaneous Biliary Intervention in Patients with Primary Sclerosing Cholangitis
Percutaneous Biliary Intervention in Patients with Primary Sclerosing Cholangitis
 Genetics of Primary Sclerosing Cholangitis
Genetics of Primary Sclerosing Cholangitis
 Cholangiocyte Biology
Cholangiocyte Biology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


