(1)
Department of Translational Medical Sciences, Section of Pediatrics, University of Naples “Federico II”, Via S. Pansini 5, 80131 Naples, Italy
Keywords
Hirschsprung’s diseaseIntestinal neuronal dysplasiaRectal suction biopsyAnorectal manometryAcetylcholinesteraseIntroduction
Hirschsprung’s disease (HD) is a heterogeneous genetic disorder, resulting from an anomaly of the enteric nervous system (ENS) of neural crest cells origin. A congenital malformation characterized by the absence of parasympathetic intrinsic ganglion cells in the submucosal and myenteric plexuses, HD is regarded as the consequence of the premature arrest of the craniocaudal migration of vagal neural crest cells in the hindgut, occurring between the 5th and 12th week of gestation, and it is therefore regarded as a neurocristopathy [1].
Epidemiology
HD occurs in approximately 1 out of 5000 live births, with a male predominance of 4:1. It is generally sporadic, although in 3–7 % of cases a genetic transmission has been reported [2]. The risk for short-segment disease is 5 % in brothers and 1 % in sisters of index cases; for long-segment disease, the risk is 10 %, regardless of sex [2].
Etiology
HD is characterized by the congenital absence of ganglion cells in the submucosal and myenteric plexuses in the distal bowel and variable proportion of the colon proximally. The embryonic disorder is a lack of the craniocaudal migration, differentiation, and maturation of neuroblasts from the neural crests, by the 5th–12th week of gestation; the earlier the migration ceases, the longer the aganglionic segment will be. The aganglionic segment is permanently contracted, causing dilatation proximal to it [3]. HD may be classified according to the length of the aganglionic segment: the classic form (short segment 70–75 % of cases) is limited to the rectum and sigmoid colon; the long segment, or subtotal colonic disease (10–15 %), generally involves the bowel up to the splenic flexure; total colonic aganglionosis (TCA) (3-6%) may extend to involve a variable amount of the short bowel; and total intestinal aganglionosis, is sometimes associated with intestinal malrotation or volvulus [2]. Ultrashort-segment aganglionosis is considered a functional alteration, without any detectable histological finding. Although longer aganglionic segments tend to produce more dramatic symptoms, some patients with even short-segment disease deteriorate rapidly [4].
Pathophysiology
The hallmark of diagnosis is the absence of ganglion cells from the myenteric and submucosal plexuses, as seen on a full-thickness or suction (mucosal–submucosal) biopsy of the rectum. Proximal contents fail to enter the unrelaxed, aganglionic segment. The lack of non-adrenergic–non-cholinergic inhibitory innervation is responsible for a tonic contraction of the affected segment, with absence of peristalsis and proximal dilation of the gut [5].
Morphologically, ganglionic cells are absent from the narrowed segment and for some distance (1–5 cm usually) into the dilated segment. The pattern of nerve fibers is abnormal also; they are hypertrophic with abundant, thickened bundles. Specific stains for acetylcholinesterase are used to highlight the abnormal morphology [6, 7].
In recent years, new insights in the pathophysiology of HD have been gained. There is a growing consensus that the disease might be the expression of a genetic alteration, as reported in the genetic section of this chapter.
It has also been suggested that abnormal expression of muscular neural cell adhesion molecule is likely to be associated with an arrest in the craniocaudal migration of neural cells to their most distal location [8]. Furthermore, the lack of nitric oxide (NO)-producing nerve fibers in the aganglionic intestine probably contributes to the inability of the smooth muscle to relax, thereby causing lack of peristalsis [9]. In addition, in the aganglionic segments, interstitial cells of Cajal (ICC) are scarce and their network appears to be disrupted [10].
Genetics
HD occurs as an isolated trait in 70 % of patients, is associated with chromosomal abnormality in 12 % of cases, trisomy 21 being by far the most frequent (> 90 %). Additional congenital anomalies are found in 18 % of cases, including gastrointestinal malformation, cleft palate, polydactyly, cardiac septal defects, and craniofacial anomalies. The higher rate of associated anomalies in familial cases than in isolated cases (39 vs. 21 %) strongly suggests syndromes with Mendelian inheritance [2]. Isolated HD appears to be a multifactorial malformation with low, sex-dependent penetrance, variable expression according to the length of the aganglionic segment, and suggesting the involvement of one or more gene(s) with low penetrance [2]. These parameters must be taken into account for accurate evaluation of the recurrence risk in relatives. Segregation analyses suggested an oligogenic mode of inheritance in isolated HD. With a relative risk as high as 200, HD is an excellent model for the approach to common multifactorial diseases [11].
A large number of chromosomal anomalies have been described in HD patients. Free trisomy 21 (Down syndrome) is by far the most frequent, involving 2–10 % of ascertained HD cases. Syndromes associated with HD can be classified as pleiotropic neurocristopathies [1], syndromes with HD as a mandatory feature, and occasional association with recognizable syndromes. The neural crest is a transient and multipotent embryonic structure that gives rise to neuronal, endocrine and paraendocrine, craniofacial, conotruncal heart, and pigmentary tissues. Neurocristopathies encompass tumors, malformations, and single- or multifocal abnormalities of tissues mentioned above in various combinations. Multiple endocrine neoplasia type 2 (MEN 2) and Waardenburg syndrome (WS) are the most frequent neurocristopathies associated with HD [12, 13].
WS, an autosomal dominant condition, is by far the most frequent condition combining pigmentary anomalies and sensorineural deafness, resulting from the absence of melanocytes of the skin and the stria vascularis of the cochlea. The combination of HD with WS defines the WS4 type (Shah–Waardenburg syndrome). Indeed, homozygous mutations of the endothelin pathway and heterozygous SRY-related box 10 (SOX 10) mutations have been identified in WS4 patients with central nervous system (CNS) involvement including seizures, ataxia, and demyelinating peripheral and central neuropathies [14].
A wide spectrum of additional isolated anomalies has been described among HD cases with an incidence of sporadic types varying from 5 to 30 % [2, 15]. No constant pattern is observed. These anomalies include distal limb, sensorineural, skin, gastrointestinal, central nervous system, genital, kidney, cardiac malformations, and facial dysmorphic features.
These data highlight the importance of a careful assessment by a clinician trained in dysmorphology for all newborns diagnosed with HD. Skeletal X ray and cardiac and urogenital echographic survey should be systematically performed. The observation of one additional anomaly to HD should prompt chromosomal studies.
Molecular Genetics
Several genes have been implicated in isolated HD; the two major ones being the proto-oncogene RET (RET) and the endothelin B receptor (EDNRB) [2].
The RET Signaling Pathway
The first observation was about an interstitial deletion of chromosome 10q11.2 in patients with TCA and mental retardation [16]. The proto-oncogene RET, identified as disease causing in MEN 2 and mapping in 10q11.2, was regarded as a good candidate gene owing to the concurrence of MEN 2A and HD in some families and the expression in neural crest-derived cells. Consequently, RET gene mutations were identified in HD patients [17]. Expression and penetrance of an RET mutation are variable and sex dependent within HD families (72 % males and 51 % females). Over 100 mutations have been identified including large deletions encompassing the RET gene, microdeletions and insertions, nonsense, missense, and splicing mutations [18–20]. Haploinsufficiency is the most likely mechanism for HD mutations. Biochemical studies showed variable consequences of some HD mutations (misfolding, failure to transport the protein to the cell surface, abolished biological activity).
Despite extensive mutation screening, an RET mutation is identified in only 50 % of familial and 15–20 % of sporadic HD case [21]. However, most families with few exceptions are compatible with linkage at the RET locus [22]. Mutations in RET ligand, like glial cell-derived neurotrophic factor (GDNF), GDNF family receptor α (GFRA)1–4, neurturin (NTN), persephin (PSPN), and artemin (ARTN), may occur, but are not sufficient to lead to HD.
The Endothelin Signaling Pathway
A susceptibility locus for HD in 13q22 was suggested for three main reasons: a significant load score at 13q22 in a large inbred Old Order Mennonite community with multiple cases of HD; de novo interstitial deletion of 13q22 in several patients with HD; and syntony between the murine locus for piebald-lethal (sl), a model of aganglionosis, and 13q22 in humans. Subsequently, an EDNRB missense mutation was identified in the Mennonite kindred (W276C) [23, 24]. Both EDNRB and EDN3 were screened in large series of isolated HD patients and EDNRB mutations were identified in approximately 5 % of the patients. It is worth mentioning that the penetrance of EDN3 and EDNRB heterozygous mutations is incomplete in those HD patients, de novo mutations have not hitherto been observed, and that S-HD is largely predominant [25].
SOX10
The last de novo mouse model for WS4 in human is dominant megalon (Dom). The Dom gene is Sox10, a member of the SRY (sex-determining factor)-like, high mobility group (HMG) DNA-binding proteins. Subsequently, heterozygous SOX10 mutations have been identified in familial and isolated patients with WS4 (including de novo mutation) with high penetrance [26].
Clinical Signs/Symptoms
The clinical symptoms of HD usually start at birth with the delayed passage of meconium. More than 90 % of term neonates, and < 10 % of children with HD, pass meconium in the first 24 h of life [27, 28]. Thus, HD must be suspected in any full-term infant who does not pass meconium in the first 24 h of life and in the premature infants who have excessively delayed the passage of meconium (7–8 days) [28]. Failure of the distal bowel to relax and allow the passage of stool leads to functional obstruction and secondary dilatation of the bowel proximal to the aganglionic segment. Affected children may present with severe dysmotility causing obstructive symptoms, ribbon-like stools, and frequently, failure to thrive. In > 90 % of affected patients, the symptoms start during the neonatal period and, in the majority, the diagnosis is made during the first 3 months of life, whereas < 1 % are diagnosed during adult life [29].
In infants and children, the presentation is often less dramatic and may not mimic acute intestinal obstruction. Severe constipation and recurrent fecal impaction are more common. Physical examination reveals a distended abdomen and a contracted anal sphincter and rectum in most children. The rectum is devoid of stool except in cases of short-segment aganglionosis. As the finger is withdrawn, there may be an explosive discharge of foul-smelling liquid stools, with decompression of the proximal normal bowel.
Complications
Over the past four decades, enterocolitis has been a major cause of morbidity and mortality in infants and children with HD. The mean incidence is 25 %, but the range is great (from 17 to 50 %) and may be differently estimated depending on the manner in which it is diagnosed. Mortality rates range from 0 to 33 %, probably reflecting differences in the diagnostic criteria [30]. Mortality also appears to be associated with other factors, such as trisomy 21. Pathogenesis is thought to be related to fecal stasis with proliferation of colonic bacteria, and, therefore, a delayed HD diagnosis seems to be a significant risk factor [31]. The classic clinical manifestations described for enterocolitis include abdominal distension, explosive diarrhea, vomiting, fever, lethargy, rectal bleeding, and shock [32]. Abdominal radiographs show the Intestinal “cutoff” sign in the rectosigmoidal region with absence of air distally. Other common findings are small bowel dilatation in 74 % and multiple air-fluid levels [33]. Because of the risk of perforation, contrast enema should not be performed in the presence of clinical enterocolitis.
Postoperative enterocolitis has been associated with fairly high rate of mortality in several series. In fact, when examining the deaths related to HD, several groups found that approximately 50 % of deaths resulted from complications directly related to an enterocolitis episode [34, 35].
Rectal washouts should be the initial approach in the care of a child, regardless of age, who presents with enterocolitis. Along with washouts, intravenous antibiotics or oral metronidazole (in mild cases) should be used. Should the disease process fail to improve, or the infants’ condition deteriorates, the performance of a leveling colostomy should be considered [34, 35].
Diagnosis
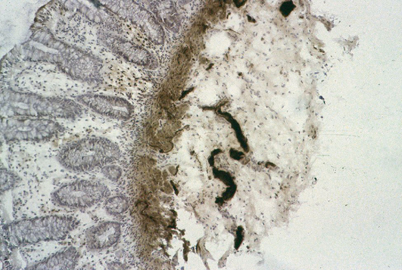
For diagnosis of HD, the subjects’ history is crucial. The main elements to obtain are the age of the appearance of symptoms, whether the passage of meconium has been normal or delayed; and whether the child presented with episodes of functional intestinal obstruction. In addition, a functional (idiopathic) megacolon must be ruled out. A clinical comparison of functional and congenital megacolon is reported in Table 22.1 [36]. When the history (early onset of constipation, absence of fecal soiling) and/or the physical examination (empty rectal ampulla) suggest an organic cause, anorectal manometry (ARM) should be performed. ARM evaluates the response of the internal anal sphincter to inflation of a balloon in the rectal ampulla [37]. When the rectal balloon is inflated, there is normally a reflex relaxation of the sphincter. The rectoanal inhibitory reflex is absent in patients with HD; there is no relaxation, or there may even be paradoxical contraction of the internal anal sphincter (Fig. 22.1) [36]. Although the absence of the rectoanal inhibitory reflex is specific for the diagnosis of HD, the role of ARM is still debated. A recent comprehensive systematic review by de Lorijn et al. [38] compared the diagnostic accuracy among rectal suction biopsy (RSB) , ARM, and Barium enema (BE) for the diagnosis of HD. Although RSB gave the highest mean sensitivity and specificity (93 and 98 %, respectively) ARM showed similar values (91 and 94 %). ARM has the advantages of being a less-invasive method without the exposure to ionizing radiation. The limitations include the need for the patient to be in a normal physiologic and quiet state to avoid possible artifacts [38, 39]. Because specificity is lower for ARM compared with RSB [38], ARM cannot reliably replace histology and biopsies. A recent practical guidelines published by ESPGHAN GI committee stated ARM should not be used as a sole diagnostic tool for HD in neonates and infants [40]; however; ARM is a useful screening test in older children presenting with chronic constipation and further symptoms suggesting HD (empty rectal ampulla, nonresponsiveness to standard therapy, early-onset constipation). If the rectoanal inhibitory reflex is absent, these patients should be referred for RSB to confirm the diagnosis of HD. If the rectoanal inhibitory reflex is present, HD could be reasonably excluded [40]. BE is helpful in the assessment of a transition zone between aganglionic and ganglionic bowel and giving an estimation of the length of an aganglionic segment . Demonstration of the transition zone is easier if no effort is made to cleanse the bowel (Fig. 22.2) [36]. In the newborn, dilatation of the proximal ganglionic bowel may not have developed, and radiological diagnosis may be more difficult. The sensitivity and specificity for recognition of a transition zone have been reported to be 80 and 76 %, respectively [41]. The BE may not show a transition zone in cases of total colonic HD, or the BE may be indistinguishable from cases of functional constipation when ultra-short-segment HD is present. Therefore, a BE should not be performed as an initial diagnostic tool because it does not represent a valid alternative to RSB or ARM to exclude or diagnose HD; however, BE may have some use as an additional investigation in diagnosed cases to assess the length of the rectosigmoid aganglionic segment before surgery [40]. Nevertheless, the diagnosis requires histological evidence. The easiest means of obtaining adequate diagnostic tissue in rectal biopsies in infants is by rectal suction biopsy [42]. An accurate diagnosis is only possible if 2–3 suction biopsies are taken 2–3 cm above the dentate line and if they include sufficient submucosa [43]. Biopsies taken closer to the dentate line may be misleading because of a normal zone of submucosal hypoganglionosis or even aganglionosis [43]. The histological diagnosis is based on the demonstration of the total absence of ganglionic cells in the affected segment of the intestine with an overgrowth of large nerve trunks in the intermuscular and submucosal zone (Figs. 22.3 and 22.4) [36]. Acetylcholinesterase (AchE) activity in normal colon shows only few fibers in the lamina propria and muscularis mucosae ; in HD, there is an increase in thick, knotted acetylcholinesterase-positive nerve fibers in the muscularis mucosae and lamina propria and hypertrophied nerve trunks in the submucosa. Techniques for histopathological diagnosis include analysis of hematoxylin- and eosin (H&E)-stained paraffin sections, snap-frozen sections stained for AchE, and immunostaining for neuronal markers. A recent consensus by Knowles et al. stated that insufficient data exist to firmly recommend one approach over the other; although, the combination of AchE and H&E staining increases the diagnostic accuracy because it improves the identification of submucosal ganglion cells, as well as demonstrates the abnormal nerve fiber distribution in the left colon [43]. The potential value of immunohistochemistry has been evaluated in a number of studies. Calretinin is an immunohistochemical marker that may be a potential alternative to AchE [44, 45]. Calretinin is normally present in the perikarya and nerve processes of a subset of enteric ganglion cells. Immunoreactivity is lost in the aganglionic segment of HD [45]. A recent comparative study shows that calretinin is as sensitive and specific as AchE [46]. However, further studies are needed to confirm calretinin immunostaining as a new standard for diagnosis .

 < div class='tao-gold-member'>
< div class='tao-gold-member'>




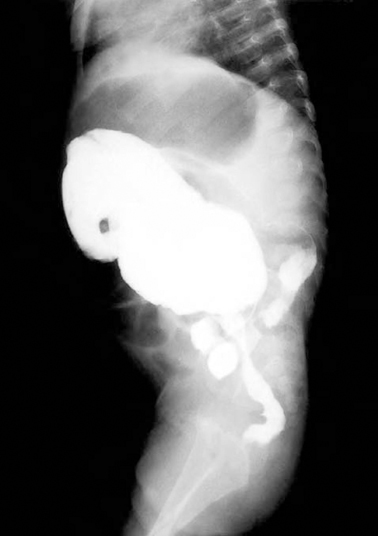
Fig. 22.1
Anorectal manometry in a 1-month-old boy with Hirschsprung’s disease. Distention of a rectal balloon with air for 1 s produces no decrease of anal pressure. (Reprinted with permission from Ref. [36], Fig. 17.2, p. 262)
Fig. 22.2
Barium enema showing a long, narrowed segment in a child with Hirschsprung’s disease. (Reprinted with permission from Ref. [36], Fig. 17.3, p. 262)
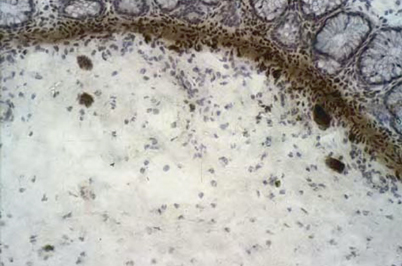
Fig. 22.3
Rectal suction biopsy in a child with functional constipation. Note the presence of clusters of neuron in the submucosa and acetylcholinesterase activity showing only few wispy fibers. (Reprinted with permission from Ref. [36], Fig. 17.4, p. 263)
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
