Fig. 4.1
A proposed multi-hit pathogenesis of IgAN. IgA1 with some O-glycans deficient in galactose (galactose-deficient IgA1; Gd-IgA1) is produced in increased amounts, leading to increased levels in the circulation (hit 1) and such molecules are recognized by unique circulating anti-glycan autoantibodies (hit 2). This process leads to formation of pathogenic immune complexes (hit 3), some of which reach the glomerular circulation and deposit in the mesangium and induce renal injury (hit 4). Moreover, there are likely upstream factors involved in abnormal mucosal immune responses characteristic for patients with IgAN
4.2 Structure of IgA1
Monomeric human IgA1 has two heavy chains and two light chains connected by disulfidic bridges [37]. Each heavy chain has three constant (Cα) domains and one variable (V) domain; each light chain has one C and one V domain (Fig. 4.2a). Unlike IgA2, the other IgA subclass in humans, IgA1 has a unique hinge region between Cα1 and Cα2 that consists of two octapeptide repeats (TPPTPSPS) and is the site of attachment of O-glycans [38]. On circulatory IgA1, usually three to six of the nine potential O-glycosylation sites are glycosylated (Fig. 4.2a) [39–42]. There are also two N-glycosylation sites on each heavy chain containing complex glycans [43–47]. Monomers of IgA1 can be covalently associated during their production in plasma cells with a ~15-kDa J-chain [48–50] and thus the immunoglobulin can be present as dimers and higher oligomers, termed polymeric IgA1. In the circulation, most of IgA1 is monomeric [51, 52].
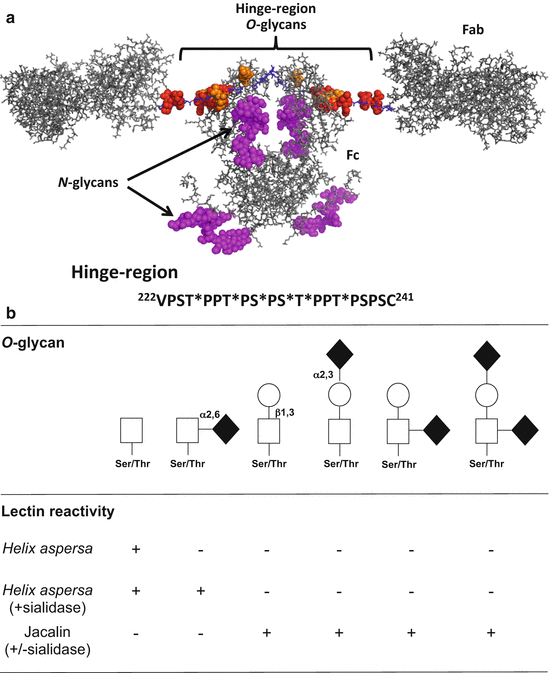
Fig. 4.2
Structure and O-glycosylation of human circulatory IgA1. (a) Modeled structure of glycosylated monomeric IgA1 based on PDB ID: 1IGA (top) and hinge-region amino-acid sequence with attachment sites of six O-glycans [125]. Modeled O– and N– glycans are depicted as spheres for clarity and are based on observed glycoforms: red for Gal-GalNAc; orange for GalNAc; and magenta for N-glycan (NeuAc)2(Gal)2(GlcNAc)2+(Fuc)1(Man)3(GlcNAc)2 [40, 46]. Hinge-region non-glycosylated amino-acid residues are in blue. There are up to six sites of O-glycosylation in the IgA1 hinge region – at T225, T228, S230, S232, T233, and T236 (marked by stars) [40]. (b) O-glycans of circulatory IgA1 and their lectin reactivities. Core 1 glycans consists of N-acetylgalactosamine and galactose, with or without sialic acid. Lectin from Helix aspersa recognizes terminal N-acetylgalactosamine whereas jacalin binds to the disaccharide composed of N-acetylgalactosamine and galactose
O-glycans of serum IgA1 of healthy individuals are usually core 1 glycans, i.e., disaccharides consisting of Ser/Thr-linked N-acetylgalactosamine with a β1,3-linked galactose (Fig. 4.2b). The disaccharide may be sialylated on either sugar or on both sugars [45, 53]. Sialic acid (in humans, N-acetylneuraminic acid) is attached to N-acetylgalactosamine of IgA1 by an alpha2,6-linkage and to galactose by an alpha2,3-linkage. Lectins (proteins that bind to particular carbohydrates) serve as useful tools to determine presence of terminal N-acetylgalactosamine or the disaccharide consisting of N-acetylgalactosamine with β1,3-linked galactose (Fig. 4.2b), but careful assessment of lectin specificities should always be performed using well-characterized IgA proteins [40–42, 54–57].
4.3 Biosynthesis of IgA1 O-Glycans
O-glycosylation of IgA1 is a stepwise process beginning with attachment of N-acetylgalactosamine to Ser/Thr residues of the hinge region; it is mediated by N-acetylgalactosaminyl-transferase 2 (GalNAc-T2) [58, 59], but other GalNAc-Ts, including GalNAc-T1, GalNAc-T11, and GalNAc-T14, may contribute to the process [60, 61]. The O-glycan chain can be then extended by attachment of galactose to the N-acetylgalactosamine residue (Fig. 4.3); this process is mediated by core 1 beta1,3-galactosyltransferase (C1GalT1) [62]. The stability of this enzyme during biosynthesis depends on interaction with its chaperone (Cosmc) for assistance with protein folding. Without this association with Cosmc, the C1GalT1 nascent protein is rapidly degraded [63–65]. The core 1 structures of IgA1 may be further modified by sialyltransferases that attach sialic acid to the galactose residues (mediated by an ST3Gal enzyme) and/or N-acetylgalactosamine residues (mediated by ST6GalNAc-II, as the usual ST6GalNAc-I is not expressed in IgA-producing cells) [61, 66, 67].
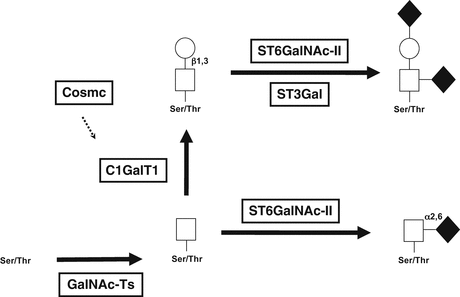
Fig. 4.3
Biosynthesis of O-glycans of IgA1. O-glycans are synthesized in a stepwise manner, starting with attachment of N-acetylgalactosamine to an oxygen molecule of serine or threonine (catalyzed by GalNAc-transferases), followed by addition of galactose (catalyzed by C1GalT1). Sialic acid can be added to each glycan by different enzymes, ST3Gal for attachment to galactose or ST6GalNAC-II for attachment to GalNAc. Sialylation of GalNAc prevents subsequent addition of galactose
4.4 Aberrant Glycosylation of IgA1 in IgA Nephropathy
Serum IgA1 of healthy individuals had been thought to contain few or no galactose-deficient O-glycans [45], but it has been shown that galactose-deficient O-glycans may be present at some sites in the IgA1 hinge region [40]. Glycosylation studies have revealed aberrancies in the O-glycans of IgA1 in the circulation of patients with IgAN. Specifically, most patients with IgAN have elevated serum levels of IgA1 with some O-glycans deficient in galactose [68–73]. Thus, a fraction of circulatory IgA1 molecules has some hinge-region O-glycans without galactose, i.e., consisting of terminal N-acetylgalactosamine or sialylated N-acetylgalactosamine (Fig. 4.2). This galactosylation defect appears to be specific for IgA1, as other O-glycosylated serum proteins such as C1 inhibitor and IgD do not exhibit galactose deficiency [40, 41, 56, 57, 74].
4.4.1 Synthesis of IgA and Possible Origin of Gd-IgA1
Humans produce approximately 70 mg of IgA per kg of body weight daily [75]. Most IgA is produced in mucosal tissues, e.g., in the gut, as polymeric IgA that is then selectively transported by a receptor-mediated pathway into the external secretions (secretory IgA); only a small percentage of polymeric IgA enters the circulation [52, 76, 77]. IgA in the circulation, predominantly of IgA1 subclass, is produced mainly in the bone marrow and to a lesser extent in the spleen and lymph nodes. A contribution of tonsillar IgA-producing cells to serum IgA has been considered to play a role in pathogenesis of IgAN [33, 34, 78–83]. IgA is rapidly catabolized; the half-life of IgA in the circulation is about 5 days. Circulatory IgA is catabolized predominantly in the liver by hepatocytes [84–92].
Genetic influences on development and expression of IgAN have been recognized and risk alleles of multiple genomic loci have been recently identified (for review, see [5, 7, 9, 93, 94]). Notably, serum levels of Gd-IgA1 are genetically determined and may be predictive of disease progression [95–98]. Thus, it is clinically relevant to determine the nature of aberrant O-glycosylation of IgA1 in IgAN at a molecular level.
Assessment of IgA1 O-glycosylation in general and in IgAN in particular initially utilized O-glycan-specific lectins and monosaccharide compositional analysis [15, 68–70, 72, 99, 100]. Lectin-based assays, using carefully selected lectins specific for particular sugars [54], evolved into a quantitative lectin ELISA [71]. This unique test has been used for analyses of serum IgA1 from multiple cohorts and the accumulated data show that most adult and pediatric patients with IgAN have elevated serum levels of Gd-IgA1 [54, 71, 73, 98, 101–105]. Moreover, the lectin-based assay was instrumental in establishing that serum levels of Gd-IgA1 are heritable in familial as well as sporadic IgAN [95].
To characterize O-glycosylation of IgA1 at a molecular level, mass spectrometric analyses have been used in addition to monosaccharide compositional analyses [16, 55, 69, 72, 106–111]. Importantly, a new approach for direct localization of O-glycan attachment sites on IgA1 has been developed by combining a technique to fragment hinge-region glycopeptides (electron-capture or electron-transfer dissociation) with high-resolution mass spectrometry, a process termed tandem mass spectrometry [40–42, 56, 57, 112]. In this approach, the individual glycoforms are identified by their molecular masses and the sites of O-glycan attachment are determined by fragmentation. These protocols revealed O-glycoform isomers, i.e., hinge-region glycopeptides with the same number of glycans but with some attached at different sites [40, 113–116]. It remains to be determined whether this microheterogeneity affects the expression or prognosis of IgAN.
4.5 Studies with IgA1-Producing Cells
Development of immortalized IgA1-secreting cells derived from cells in the circulation of patients with IgAN and healthy and disease controls provided a new tool for studies of normal and aberrant O-glycosylation of IgA1 [103]. IgA1 secreted by the cells from patients with IgAN had higher degrees of galactose deficiency of the O-glycans than that from cells of healthy controls. Notably, the relative degree of galactose deficiency of the IgA1 secreted by IgA1-producing cells correlates with that of serum IgA1 from the corresponding donor [103]. Thus, these cells served as an excellent model system to study enzymatic pathways and the actual O-glycosylation of secreted IgA1. Initial studies showed that the polymeric IgA1 form is the most affected by galactose deficiency. Furthermore, this phenotype is related to decreased expression and activity of C1GalT1 and elevated expression and activity of ST6GalNAc-II [103]. Moreover, the expression of C1GalT1-specific chaperone Cosmc [63, 64], that is necessary for stability of the nascent C1GalT1 protein, is decreased [103]. More recently, the roles of C1GalT1 and ST6GalNAc-II in production of Gd-IgA1 have been confirmed, using siRNA knockdown and biochemical approaches [67, 117, 118].
IgA1 secreted by our established cell lines was isolated and its O-glycosylation characterized by using high-resolution mass spectrometry. The IgA1 secreted by cells from patients with IgAN has more galactose-deficient O-glycans, consisting of terminal or sialylated N-acetylgalactosamine, compared to IgA1 from the cells from healthy controls [119]. Moreover, IgA1 from patients with IgAN also had more O-glycans per heavy chain. Together, these data revealed another type of glycosylation abnormality, an elevated number of O-glycans on IgA1 from patients with IgAN. This finding implicated a central role for an O-glycosylation-initiating enzyme, a GalNAc-T, in production of Gd-IgA1.
Several GalNAc-Ts are abundantly expressed in IgA1-producing cells [61, 120], including GalNAc-T2 [103]. Of this family of enzymes, only GalNAc-T14, the closest structural relative of GalNAc-T2, was expressed at several-fold greater levels in the cells from patients with IgAN compared to cells from healthy controls [120]. Conversely, the expression of GalNAc-T2 and other GalNAc-Ts did not differ between patients and healthy controls [103]. These findings thus indicate that overexpression of GalNAc-T14 may contribute to enhanced O-glycosylation of IgA1 in IgAN [9, 61].
Notably, the dysregulated expression and activities of several key glycosyltransferases are further enhanced by some cytokines, including IL-6 [117]. The signaling process through the IL-6 receptor/gp130 complex is mediated by STAT3 [121, 122] and increases activity of ST6GalNAc-II and decreases activity of C1GalT1 (Fig. 4.4) [117]. This process leads to greater production of Gd-IgA1 [117]. Another type of signal involved in cellular differentiation and survival that may be relevant to Gd-IgA1-producing cells includes B-cell activating factor (BAFF) [122]. Notably, overexpression of human BAFF in mice leads to overproduction of IgA and development of IgA glomerular deposits [123].
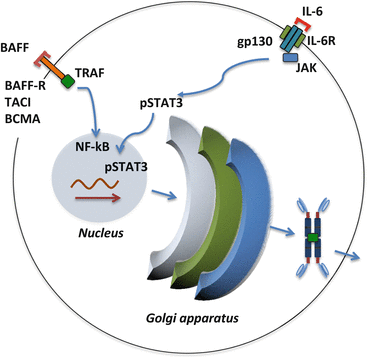
Fig. 4.4
Examples of two signaling pathways involved in cell survival and differentiation of IgA1-producing cells. BAFF is a growth/differentiation factor for plasma cells and its overproduction may enhance survival or differentiation of cells involved in production of autoantigen, Gd-IgA1. IL-6 can increase production of Gd-IgA1 in IgA1-secreting cells of patients with IgAN
These observations may together explain why the clinical onset or a flare in the activity of IgAN often coincides with an active mucosal infection of the upper respiratory tract and/or digestive system. It can be speculated that such environmental factors exert their activity through cytokines (e.g., IL-6) and growth factors (e.g., BAFF, APRIL) (Fig. 4.4) to upregulate cellular production of Gd-IgA1 and/or cell survival in susceptible individuals [9, 94, 122].
4.6 Summary and Implications for Diagnosis, Prognosis, and Treatment of IgA Nephropathy
Multiple lines of evidence allow us to characterize IgAN as an autoimmune disease in which Gd-IgA1, produced in increased amounts and leading to increased levels in the blood, is bound by unique autoantibodies. This sequence of events leads to formation of pathogenic circulating Gd-IgA1-containing immune complexes. Some of these complexes deposit in the kidney and induce a mesangioproliferative glomerular injury [124]. Elevated serum levels of Gd-IgA1 are genetically codetermined and can be further increased by some cytokines. Moreover, serum levels of Gd-IgA1 may be predictive of disease progression. Gd-IgA1 is produced by IgA1-secreting cells of patients with IgAN due to dysregulation of several key glycosylation enzymes. A combination of approaches with use of IgA1-producing cell lines and high-resolution mass spectrometry will likely provide a better understanding at cellular and molecular levels of the heterogeneity of Gd-IgA1 in patients with IgAN. We believe that through a detailed understanding of the disease processes, biomarkers specific for IgAN can be identified and developed into clinical assays to aid in the diagnosis, assessment of prognosis, and monitoring of the disease progression. Moreover, characterization of specific pathogenetic pathways, such as those involved in Gd-IgA1 production, will identify targets for future disease-specific therapies.
Acknowledgments
This review is dedicated to our late colleague, Dr. Milan Tomana, a pioneer in the studies of IgA glycosylation and the abnormalities associated with IgAN. The authors also thank all coworkers and collaborators who have participated in various aspects of these studies. The authors have been supported in part by grants DK078244, DK082753, DK099228, DK105124, DK106341, DK079337, and GM098539 from the National Institutes of Health and a gift from the IGA Nephropathy Foundation of America.
References
1.
Berger J, Hinglais N. Les dépôts intercapillaires d’IgA-IgG (intercapillary deposits of IgA-IgG). J Urol Nephrol. 1968;74:694–5.
2.
D’Amico G, Colasanti G, Barbiano di Belgioioso G, Fellin G, Ragni A, Egidi F, et al. Long-term follow-up of IgA mesangial nephropathy: clinico-histological study in 374 patients. Semin Nephrol. 1987;7(4):355–8.PubMed
3.
Julian BA, Waldo FB, Rifai A, Mestecky J. IgA nephropathy, the most common glomerulonephritis worldwide. A neglected disease in the United States? Am J Med. 1988;84:129–32.PubMed
4.
D’Amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol. 2004;24(3):179–96.PubMed
6.
Beerman I, Novak J, Wyatt RJ, Julian BA, Gharavi AG. Genetics of IgA nephropathy. Nat Clin Pract Nephrol. 2007;3:325–38.PubMed
7.
Kiryluk K, Julian BA, Wyatt RJ, Scolari F, Zhang H, Novak J, et al. Genetic studies of IgA nephropathy: past, present, and future. Pediatr Nephrol. 2010;25(11):2257–68.PubMedPubMedCentral
8.
Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765.PubMedPubMedCentral
9.
Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest. 2014;124(6):2325–32.PubMedPubMedCentral
10.
Julian BA, Quiggins PA, Thompson JS, Woodford SY, Gleason K, Wyatt RJ. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N Engl J Med. 1985;312:202–8.PubMed
11.
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43:321–7.PubMedPubMedCentral
12.
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46(11):1187–96.PubMedPubMedCentral
13.
Julian BA, Suzuki H, Suzuki Y, Tomino Y, Spasovski G, Novak J. Sources of urinary proteins and their analysis by urinary proteomics for the detection of biomarkers of disease. Proteomics Clin Appl. 2009;3(9):1029–43.PubMedPubMedCentral
14.
Jennette JC. The immunohistology of IgA nephropathy. Am J Kidney Dis. 1988;12(5):348–52.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Differences in Etiology and Treatment in Scandinavian Countries
Advances in Genetics of Immunoglobulin A Nephropathy
Differences in Etiology and Treatment in Scandinavian Countries
Advances in Genetics of Immunoglobulin A Nephropathy
 How Different Are the Current Understandings of Treatments for IgA Nephropathy?
How Different Are the Current Understandings of Treatments for IgA Nephropathy?
 Podocyte Injury and Significance of Urinary Podocalyxin and Megalin
Podocyte Injury and Significance of Urinary Podocalyxin and Megalin
 IgA Nephropathy from the VALIGA European Study: Differences in Treatment Approaches Within Europe
IgA Nephropathy from the VALIGA European Study: Differences in Treatment Approaches Within Europe
 Complement Activation
Complement Activation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


