Idiopathic
Genetic
• PRSS1
• CFTR
• SPINK1
• CTRC
• CPA1
• CEL
Drugs (l-asparaginase, valproate, metronidazole, azathioprine, tetracycline, pentamidine, etc.)
• Metabolic disease
• Hyperlipidemia
• Hypercalcemia
• Glycogen storage disease
• Organic acidemias
Autoimmune
Anatomic
• Pancreas divisum
• Anomalous junction of the biliary and pancreatic ducts
• Annular pancreas
• Ampullary obstruction
• Crohn’s disease
Pathophysiology
The pathophysiology of CP is incompletely understood. The pancreas is not easily accessible for sampling; therefore, it is difficult to detect the earliest changes and identify the events that contribute to the pathophysiology of CP. In children with hereditary pancreatitis (HP) , mutations in the cationic trypsinogen (PRSS1) gene cause activation of trypsinogen within the acinar cells as an early event in pancreatitis [19]. Premature activation of trypsinogen to trypsin initiates an activation cascade, causing additional trypsinogen activation and conversion of other digestive proenzymes to active enzymes, leading to pancreatic digestion and inflammation [11].
A widely accepted hypothesis is that CP begins with an episode of acute pancreatitis (AP), followed by an ongoing chronic or recurrent inflammation that leads to fibrotic replacement of acini and islet cells [1]. In the sentinel acute pancreatitis event (SAPE) hypothesis [20], a metabolic or oxidative stress initiates the first episode of AP (the sentinel event). Activated lymphocytes, macrophages, and stellate cells increase in number within the pancreas, and then produce cytokines and deposit small amounts of collagen. Most patients recover uneventfully from AP, and the pancreas returns to normal. In some patients, due to the continued presence of stress, inflammatory cells and stellate cells remain active, release cytokines, and deposit collagen, eventually producing the fibrotic changes characteristic of CP. Although the process may be started and perpetuated by environmental factors, other factors (i.e., genetic) must be present for CP to develop in some individuals and not others.
Diagnosis
Clinical Presentation
Abdominal pain is the most common symptom of CP. Pain is usually in the upper abdomen, episodic or persistent, mild–moderate to severe. Older children describe the pain as deep and penetrating, radiating to the back, and worse after meals. Younger children cannot verbalize the pain well, so clinicians must have a high index of suspicion in this age group [21]. Commonly, younger children may be misdiagnosed as having other gastrointestinal (GI) illnesses, such as gastroesophageal reflux, constipation, or functional abdominal pain . Nausea and/or vomiting, anorexia, weight loss may be present.
Children with HP usually present with recurrent episodes of abdominal pain and symptom-free intervals [11]. The acute attacks may be triggered by a fatty meal, stress, or environmental factors such as infection, smoking, and alcohol use. Median age of presentation for HP is 10 years of age, and in 50 % of patients, CP develops 10 years after the first bout of AP [22]. Some patients may present with CP without a clear history of AP [23].
The pancreas has extraordinary reserves and does not manifest signs of insufficiency until lipase secretion is reduced to less than 5 % of the maximum output [24]. In adults, exocrine pancreatic insufficiency (EPI) occurs in 50–80 % of patients in a median time of 5.6–13.1 years [25, 26].
Diabetes mellitus can also occur in 40–70 % of adults with CP with a median time to onset of 11.9–26.3 years [25, 26]. The exact mechanism of pancreatic diabetes is unknown, but islet destruction is a late phenomenon. The exocrine and endocrine insufficiencies in pediatric CP are also incompletely studied.
If EPI has developed, symptoms of maldigestion/malabsorption, such as weight loss and steatorrhea, may be present. Fat-soluble vitamin deficiencies (A, D, E, K) can be seen. Patients with extrahepatic biliary obstruction from fibrosis in the head of the pancreas or from the pseudocyst can have jaundice. Physical examination is usually normal.
Differential Diagnoses
The differential diagnosis of CP includes causes of recurrent and chronic abdominal pain in childhood, such as peptic ulcer disease , gastritis , gallbladder disease, intestinal obstruction, Crohn’s disease, functional abdominal pain , lactose intolerance, and constipation. For the differential diagnosis of malabsorption in the presence of EPI , one should consider severe enteropathies, cholestatic liver disease , celiac disease, cystic fibrosis (CF), and rare isolated pancreatic enzyme deficiencies.
Diagnostic Testing
The INSPPIRE ( INternational Study Group of Pediatric Pancreatitis: In search for a cuRE) definitions of CP require one of the following: (1) abdominal pain consistent with pancreatic origin and imaging findings suggestive of chronic pancreatic damage, (2) evidence of EPI and imaging findings suggestive of pancreatic damage, or (3) evidence of endocrine pancreatic insufficiency and imaging findings suggestive of pancreatic damage, or (4) a surgical or pancreatic biopsy demonstrating histopathology compatible with CP [15] (Table 34.2).
Table 34.2
Diagnostic tests in chronic pancreatitis
Laboratory tests (serum amylase, lipase, Ca, lipid panel, total and direct bilirubin, alkaline phosphatase, GGT, AST, ALT, fasting serum glucose) |
Fecal elastase 1 |
72 h fecal fat |
Sweat Cl/NPD |
Imaging studies (US, CT, MRCP, EUS, ERCP) |
Genetic testing (PRSS1, CFTR, SPINK1, CTRC) |
These lab tests and abbreviations look out of place. May be they should go to a an abbreviation section. According to INSPPIRE criteria, suggestive imaging findings of CP or chronic pancreatic damage include:
Ductal changes: irregular contour of the main pancreatic duct or its radicals; intraductal filling defects; calculi, stricture, or dilation;
Parenchymal changes: generalized or focal enlargement, irregular contour (accentuated lobular architecture), cavities, calcifications, heterogeneous echotexture.
Imaging modalities may include CT, MRI/magnetic resonance cholangiopancreatography (MRCP), endoscopic retrograde cholangiopancreateography (ERCP); US; endoscopic US (EUS ; in which at least five EUS features (as defined by the Rosemont Classification [29] must be fulfilled)).
EPI is recommended to be diagnosed via fecal elastase-1 monoclonal assay < 100 mcg/g stool (two separate samples done ≥ 1 month apart) or coefficient of dietary fat absorption < 90 % on a 72-h fecal fat collection. Neither test should be performed during an AP episode, as the results may be temporarily low.
Endocrine pancreatic insufficiency can be diagnosed via 2006 WHO criteria for the diagnosis of diabetes mellitus (fasting glucose ≥ 7.0 mmol/L (126 mg/dL) or plasma glucose ≥ 11.1 mmol/L (200 mg/dL) 2 h after glucose load 1.75 g/kg children (to maximum 75 g glucose load)) [30].
Biochemistry
There are no specific laboratory tests for CP. In most cases, serum amylase and lipase are normal or only mildly elevated [31]. Aspartate transaminase (AST), alanine transaminase (ALT), GGT, alkaline phosphatase, and direct bilirubin are useful to rule out a biliary obstruction.
Radiology
Diagnostic imaging plays an important role in the initial diagnosis of CP and further planning for endoscopic and surgical interventions. The imaging modalities most commonly used in pediatric pancreatitis are summarized below.
Ultrasonography
US is usually the first imaging modality when pancreatitis is suspected in children. Smaller size of patients, lack of fat, and prominence of the left hepatic lobe make US of the pancreas more feasible in children than in adults [32]. US is 50–80 % sensitive in diagnosing CP in adults; the diagnostic accuracy of US in pediatric CP has not been studied [4]. US is most helpful in assessing the pancreatic duct diameter in children with CP (normals are ≤ 1.5 mm in children 1–6 years; ≤ 1.9 mm at ages 7–12 years; ≤ 2.2 mm at ages 13–18 years). Calcifications of the pancreas and intraductal stones can also be observed with US in CP [32].
Endoscopic US
The high diagnostic accuracy and the low complication rate of EUS (1 %) compared to ERCP have been the driving forces for the use of EUS [32]. EUS is technically feasible in children as young as 5 years of age [33], but most of the experience comes from the adult literature, and very little is written in pediatrics [34]. EUS is highly operator dependent, and even in adults there is controversy regarding its accuracy and reliability. EUS images are commonly scored based on the presence of parenchymal and ductal features. An expert consensus conference was convened in 2009 to address the controversies and developed the Rosemont classification [29]. The Rosemont criteria for CP diagnosis have not been validated in children.
Magnetic Resonance Cholangiopancreatography
MRCP can visualize the pancreas, pancreatic ducts, and the pancreaticobiliary tree and reliably detect pancreas atrophy, ductal dilatations, small filling defects, strictures, irregularities of the main pancreatic duct, and irregularity (sacculation and/or ectasia) of side branches [35] (Fig. 34.1). Due to its noninvasive nature and lack of radiation, MRCP has become the diagnostic test of choice in children with CP [36, 37]. Unlike ERCP that images ducts under pressure, MRCP visualizes the ducts in their normal physiologic state [32]. Therefore, MRCP may not reveal the details of small ducts, which may be important in diagnosing early CP.
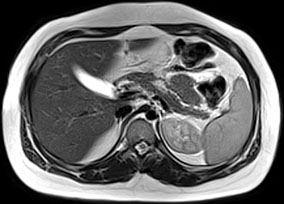
Fig. 34.1
MRCP changes in chronic pancreatitis. T1-weighted image shows atrophy of the parenchyma in a child with idiopathic chronic pancreatitis (arrows). (Photo courtesy of Dr. Simon C. Kao, University of Iowa, USA)
Secretin induces fluid secretion in the pancreatic duct, and when administered with MRCP, it increases the diameter of pancreatic duct up to 3 mm in 3–5 min with progressive decline to baseline in 10 min. Secretin may be more important in children than in adults as it increases the detectability of the normally smaller pancreatic ducts. In a pediatric study, secretin increased the number of main pancreatic duct segments visualized on MRCP from 53 to 93 %; the visualization of the duct of Santorini increased from 7 to 53 % and the detection of side branches increased from 20 to 47 % [38]. Secretin may also increase the number of false-positive reports [39]. In one pediatric study, the pancreatic diameter increase after secretin was small and did not have any impact on image quality or duct visibility of the MRCP [40]. More studies with secretin-enhanced MRCP are needed to better understand its diagnostic accuracy in pediatric CP.
Endoscopic Retrograde Cholangiopancreateography
With MRCP being widely available, ERCP is now mainly reserved for therapeutic interventions (pancreatic duct stenting, sphincterotomy, stone extraction). ERCP carries an overall morbidity of ~7 %, which includes AP (4 %) , hemorrhage (1 %), cholangitis (1 %), perforation (0.5 %), and death (0.1 %) [41]. In CP, ERCP findings include main pancreatic duct dilatation, ductal stones, and changes in the main duct branches and small ducts (Fig. 34.2). ERCP is no longer an attractive first-line test because of its potential complications and low sensitivity for detecting early-stage CP.
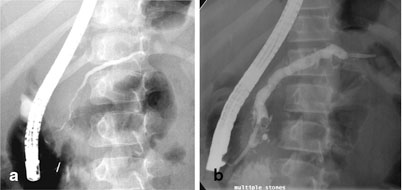
Fig. 34.2
ERCP changes in chronic pancreatitis. a Dilatation of the mid and distal portions of the pancreatic duct in a child with idiopathic chronic pancreatitis (arrows). b Dilatation of pancreatic duct with multiple filling defects (stones, arrowheads) in the distal pancreatic duct in a child with hereditary pancreatitis (R122H mutation). A pancreatic duct stent (arrow) has been placed to bypass the obstruction. (Photo courtesy of Dr. Simon C. Kao, University of Iowa, USA)
An ERCP-based grading system of CP has been developed in adults (Cambridge classification) for the classification of disease severity. Changes are graded as normal, equivocal, mild, moderate, and severe, based on the appearance of the main pancreatic duct and side branches on the ERCP. Interpretation is subjective and there can be substantial inter- and intraobserver variation. The Cambridge classification has not been validated in children [42].
Computerized Tomography (CT) with Contrast
CT can detect advanced changes in CP, such as calcification, gland atrophy, fat replacement, and ductal dilatation. The advantage of CT is that it can evaluate pancreas for other pathology and detect other causes of chronic abdominal pain. CT has poor sensitivity to identify ductal abnormalities and subtle parenchymal changes; high radiation dose is another drawback [4, 43].
Genetic Testing
Genetic testing is indicated as a diagnostic test in children with CP to determine the underlying cause. Predictive testing is not recommended in individuals without pancreatic disease and a positive family history of pancreatitis. The presence of PRSS1 mutation provides an adequate etiology for pancreatitis, but the presence of CFTR or SPINK1 mutations should not preclude a careful search for additional etiologies.
Cationic Trypsinogen (PRSS1)
UniGene name: protease, serine 1: PRSS1 gene was the first pancreatitis-specific susceptibility gene identified in families with HP [19] . In the physiological state, trypsinogen is converted to trypsin in the duodenum by the brush border-derived enterokinase. Trypsin will then activate other proteases , which will initiate intraluminal digestion of nutrients. A total of 80–90 % of individuals with HP, an autosomal dominant disease with incomplete penetrance, carry R122H mutation [19, 22]. This mutation is caused by the substitution of the amino acid histidine (H) for arginine (R) at position 122 (R122H) of the cationic trypsinogen gene. Another mutation, N29I, is caused by substitution of isoleucine (I) for asparagine (N) at position 29. R122H and N29I are the most common mutations seen in patients with HP [44]. Genetic defects in PRSS1 result in gain of function either by increasing activation or by preventing inactivation of trypsin, leading to pancreatic autodigestion .
Genetic testing for PRSS1 mutations is recommended in patients with recurrent attacks of AP and CP of unknown etiology or a positive family history of pancreatitis in first-degree or second-degree relatives.
CFTR
Pancreatitis occurs in 2–4 % of all patients with CF and 15–20 % of pancreatic-sufficient patients with mild CFTR mutations during adolescence or later in life [45, 46]. Pancreatitis is thought to result from thick secretions causing pancreatic ductular obstruction, ineffective clearing of secretions from the pancreatic duct, and autodigestion of the pancreas by activated proteolytic enzymes [11].
Patients with idiopathic CP carry a higher frequency of CFTR mutations (~ 10 %) than the general population, and a subset of these patients have CF (~ 20 %) [47–50]. CFTR mutations may also contribute to the development of CP in patients who have additional risk factors for CP [51].
There are over 1900 CFTR mutations, and only a small percentage are CF-causing mutations.
The majority of mutations (~ 40 %) have unknown functional consequences, others confer less severe disease, and the rest are considered to be benign polymorphisms with no pathogenic potential [52, 53]. Therefore, in a patient with recurrent attacks of AP or CP and CFTR mutations, sweat test is still the first test to order. A referral to a CF clinic can be made if sweat chloride is borderline (40–59 mmol/L) or abnormal ( ≥ 60 mmol/L). Nasal potential difference (NPD) testing can also be helpful in measuring CFTR channel function in patients with recurrent attacks of pancreatitis [54].
SPINK1
Synthesis of PSTI, which is coded by the SPINK1 gene, protects against premature activation of trypsin within the pancreas before it is secreted into the duodenum [20]. Mutations in the SPINK1 gene (mainly N34S mutation, substitution of asparagine by serine at codon 34 in exon 3) increase the susceptibility to AP and predisposes to CP [55, 56]. Interestingly, the risk of an asymptomatic SPINK1 carrier developing pancreatitis is thought to be only 1 %. Therefore, SPINK1 mutations alone are not sufficient to cause pancreatitis, but may act as genetic modifiers in initiating the development of pancreatitis [56].
Chymotrypsin-C
Exocrine Pancreatic Function Testing
These tests can detect CP in its most advanced stage, when EPI has developed. They are not specific for CP as they can be abnormal in patients with other causes of EPI (CF, Shwachman–Diamond syndrome, etc) .
Pancreatic Stimulation Test
This test involves the collection of pancreatic fluid secreted into the duodenum and measurement of fluid volume, pancreatic enzymes, and electrolytes before and after stimulation with cholecystokinin (CCK) and secretin [60]. Although it is considered “gold standard” to quantify the exocrine pancreatic function, stimulation test is not widely performed because of its invasive nature. The collection of the duodenal fluid via the endoscope (endoscopic pancreatic function test or ePFT) has been proposed as an alternative, but this approach may underestimate the pancreatic secretary capacity and classify patients as pancreatic insufficient erroneously [60].
72-h Fecal Fat Collection
This test relies on the exocrine pancreas losing greater than 95 % of its enzyme secretory output and development of steatorrhea [24]. Steatorrhea can be measured by a 72-h stool collection and calculation of coefficient of fat absorption (CFA: (grams of fat ingested-grams of fat excreted)/(grams of fat ingested) × 100). In children younger than 6 months of age, a fecal fat greater than 15 % of fat intake is considered abnormal; this value is 7 % for children over 6 months of age. This test is not very reliable and reproducible. It is not a popular test by the families or the technical staff. The sample and the data collection are not always accurate.
Fecal Elastase-1 (FE1)
This enzyme-linked immunosorbent assay (ELISA)-based method is easy to use, cheap, and now the preferred test to diagnose EPI. A value of less than100 µg/g is considered diagnostic of EPI. Intermediate values of fecal elastase (100–200 µg/g) may be due to loss of pancreatic function, but not severe enough to cause clinical EPI. The sensitivity of FE1 to diagnose moderate and severe EPI is close to 100 %. In patients with mild loss of pancreatic function, the test sensitivity is ~25 % with a specificity of 96 % [61]. Therefore, the value of FE1 to determine patients with mild EPI or borderline normal pancreatic function is limited. FE1 may be falsely low when the stool is diluted as a result of infectious diarrhea, severe enteropathies, short gut, or if it is collected from an ileostomy.
Secretin MRCP
The pancreatic exocrine function can be assessed by the duodenal filling, changes in pancreatic duct caliber, change in anteroposterior diameter of the pancreas, and change in signal intensity ratio between pancreas and spleen on T1-weighted and arterial–venous enhancement ratios [62, 63]. The results show correlation with other exocrine pancreatic tests including ePFT [63] and fecal elastase [64]. The test has not been validated in children.
Treatment
Medical Treatment
For children presenting with an acute attack of pancreatitis, we recommend conservative management, withholding food and drink for a few days, and offering pain control. In patients with advanced disease, the treatment is directed at the complications, including chronic pain, EPI, and diabetes mellitus.
Pain Control
Analgesic medications are the mainstay of pain management. There are no prospective therapeutic trials of pain management in children; thus, the practice is guided by common practice and expert opinion. Nonsteroidal anti-inflammatory drugs and acetaminophen are the first-line agents for pain control. Long-acting and short-acting narcotics can be used in a stepwise approach. The addictive profile of narcotics and their GI side effects should be considered when initiating the therapy. Tramadol and gabapentin have shown efficacy in controlling pain in adults with CP [65].
Pancreatic Enzyme Supplementation
Pancreatic enzymes are often prescribed theoretically to reduce the feedback loop in the duodenum by reducing the CCK release and inhibiting pancreatic exocrine activation . Current evidence on the effect of pancreatic enzymes on pain control is conflicting because of methodological issues and types of preparation (enteric-coated vs. nonenteric-coated tablets) [4]. Some early studies reported benefit with nonenteric-coated preparations [66, 67], while enteric-coated preparations did not work [68, 69] possibly because the release of nonenteric-coated enzymes and feedback inhibition of pancreatic enzymes were more proximal than distal in the intestine. A recent Cochrane review evaluated the efficacy of pancreatic enzymes in patients with CP [70]. Although some individual studies reported a beneficial effect of pancreatic enzyme over placebo in improving pain, incidence of steatorrhea, and analgesic consumption, the results of the studies could not be pooled for these outcomes. Overall, the role of pancreatic enzymes in CP was reported to be equivocal .
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


