ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS AND TREATMENT
Hemochromatosis classically refers to HFE-mediated genetic iron overload, but several alternatively mediated genetic iron overload syndromes have been described.
C282Y is the major mutation and H63D the minor mutation of the HFE gene; individuals with two copies of C282Y or one copy of both mutations (compound heterozygote) are at risk for iron overload.
Excess iron deposition in tissues leads to end-organ damage; advanced hemochromatosis typically involves the liver first and may also involve the pancreas, heart, pituitary gland, and other organs.
Most patients are identified by laboratory screening or family history and are asymptomatic at diagnosis.
Symptomatic patients usually present with nonspecific complaints of fatigue, arthralgias, and abdominal pain.
Screening studies include serum iron/total iron-binding capacity (abnormal if >45%) and serum ferritin (abnormal if >200 μg/L in women, >250 μg/L in men); if either test is positive, genetic testing should be pursued.
Liver biopsy to assess iron concentration and hepatic iron index is indicated if serum ferritin is >1000 μg/dL or liver tests are abnormal.
Early diagnosis (precirrhotic stage) and treatment is associated with a normal life expectancy.
GENERAL CONSIDERATIONS
Eighty percent of clinically established cases of hemochromatosis worldwide and over 90% of cases in the United States result from the autosomal-recessive inheritance of two copies of the major mutation (C282Y) of the HFE gene. Individuals with this genotype are described as C282Y homozygotes. Ten to 15% of Caucasian populations are heterozygotes, possessing one copy of the major mutation. This mutation is believed to have originated more than 6000 years ago amid Celtic or Viking ancestry. It has been postulated that the mutation may have had a potential selective advantage, preventing iron deficiency in the setting of scarce resources such as red meat.
Prevalence estimates of homozygosity for the C282Y mutation in populations of Northern European descent are 1 in 260 persons. Prevalence rates vary by gender and ethnicity. The disease is more common in men, which is attributed to increased dietary iron consumption as well as iron losses in women during menstruation. The disease is rarely seen in African Americans, Mexican Americans, and Asians. Approximately 90–95% of HFE-related hemochromatosis cases in the United States are related to homozygosity for C282Y and 80% in European populations (EASL p. 4).
One copy of the H63D mutation, the minor mutation of the HFE gene, may be found in 15–40% of Caucasian populations. About 4% of cases of hemochromatosis result from the inheritance of one copy of the major mutation, C282Y, and one copy of the minor mutation, H63D. These individuals are described as compound heterozygotes. However, the great majority of compound heterozygotes will not develop clinically significant iron overload. The risk has been estimated to be 200 times less than in C282Y homozygotes. H63D homozygotes typically do not develop clinically significant iron overload. Individuals with this genotype may have an elevated transferrin saturation, but an elevated ferritin should prompt a search for secondary causes of iron overload.
Phenotypic expression of these altered gene states is variable and, as a result, the proportion of patients who go on to develop clinically significant hemochromatosis remains uncertain. It has been estimated that 38–50% of C282Y homozygotes will develop iron overload, but only 10–33% will manifest hemochromatosis-related morbidity. Men are much more likely than women to develop iron overload-related disease (28% vs 1%). The majority of individuals who develop end-organ damage likely have cofactors such as significant alcohol intake or nonalcoholic steatohepatitis (NASH). Overall, secondary causes of iron overload are still more common than primary iron overload syndromes.
[PubMed: 18199861]
[PubMed: 11529872]
PATHOGENESIS
The quantity of total body iron is closely regulated and is estimated to be approximately 3 g in women and 5 g in men. Most of this iron is incorporated into red blood cells. The remainder is stored in the liver and a small amount in skeletal muscle. Almost all iron absorption occurs in the duodenum. After iron is absorbed, it circulates bound to the carrier protein transferrin for distribution to tissues. In addition to taking up inorganic iron in this way, the duodenal enterocytes may also take up iron in the form of heme.
Most of the circulating iron is taken up within sites of erythropoiesis, where it is incorporated into hemoglobin by developing erythrons. The liver serves as a storage reservoir for iron and then releases iron back into circulation as needed. As red blood cells senesce, they are phagocytosed by macrophages which then process and release iron for recycling into newly developing cells at a controlled rate.
The amount of iron absorbed from food can be upregulated quickly when excess iron is lost or utilized, such as through menstruation, pregnancy, or gastrointestinal bleeding. Small amounts of iron, on the order of 1–2 mg daily, are lost as cells of the gastrointestinal and urogenital tracts are shed. An additional 1–2 mg of iron is lost daily by women during their reproductive years. However, the human body has no effective physiologic mechanism for excreting excess iron. The duodenal enterocytes must correctly sense or be signaled to absorb enough iron to replace losses but no more.
The duodenal enterocytes, hepatocytes, and macrophages all appear to play important roles in iron homeostasis. Because they function at sites distant from one another, it has been hypothesized that they communicate via a hormone or hormones. Multiple factors influence iron absorption, including both systemic and intestinal factors. Systemic factors include the level of body iron stores, erythropoietic activity, hemoglobin concentration, and oxygen saturation as well as the presence or absence of inflammatory cytokines. Intestinal factors include pancreatic insufficiency and disturbances in pH. Disorders of iron metabolism develop when disease states overwhelm the homeostatic mechanisms. For example, infections or chronic inflammation induce iron sequestration by macrophages and signal a decrease of iron absorption, leading to the anemia of chronic disease. Conversely, disorders of erythropoiesis, such as the thalassemias, release signals promoting iron absorption by developing erythrons that overwhelm the inhibitory signals generated by excessive accumulation of iron stores.
[PubMed: 15654801]
In hereditary hemochromatosis (HHC), one or more genetic mutations lead to excess iron absorption relative to body iron stores. The excess iron is stored in the liver initially but, if unrecognized and untreated, the iron may deposit in multiple end organs when hepatic storage is saturated, leading to the phenotypic expression of the disease. Iron spillover has been observed once total body stores reach 4.5 g with hepatic levels in excess of 400 μg/g of tissue.
In 1996, Feder and colleagues published the discovery of a candidate gene for hemochromatosis located on the short arm of chromosome 6, now called HFE. As noted earlier, two mutations are now recognized. The major mutation results from a tyrosine substitution for cysteine at the 282 amino acid position on the gene and is abbreviated as Cys282Tyr or simply C282Y. The minor mutation results from an aspartate substitution for histidine at the 63rd amino acid position and is abbreviated as His63Asp or simply H63D.
[PubMed: 8696333]
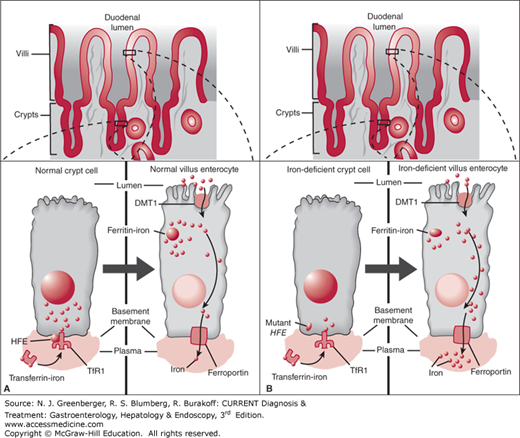
The initial model for iron overload in HFE-related hemochromatosis postulated that the HFE mutation acted at the level of the developing duodenal crypt cell. These cells elaborate iron transporters (DMT-1), as they differentiate and migrate toward the villus tip (Figure 42–1). Within this so-called crypt-programming model, the crypt cells would correctly assess or “sense” the amount of iron in circulation through HFE-dependent uptake of transferrin-bound iron at the basolateral membrane and thereby program production of the appropriate number of iron transporters in the differentiating cell. The HFE mutation would disable normal uptake of iron by the nascent crypt cells, leading to incorrect “sensing” of an iron-deficient state and to programming of an excessive number of iron transporters (Figure 42–2).
The crypt-programming model has been challenged by more recent experimental data. Changes in iron absorption occur within hours of a change in iron status, whereas enterocyte maturation takes days. Furthermore, other forms of iron overload, such as juvenile hemochromatosis, occur in the absence of HFE mutations. This suggests the existence of other factors with a more fundamental role in iron homeostasis.
Figure 42–1.
Hepcidin model. Hepcidin normally functions as a regulatory protein, inhibiting release of iron from villus enterocytes and macrophages, which in turn inhibits iron absorption. A. Normal. B. HFE-related hemochromatosis. (Adapted, with permission, from Pietrangelo A. Hereditary hemochromatosis—a new look at an old disease. N Engl J Med. 2004 Jun 3;350(23):2383–2397.)
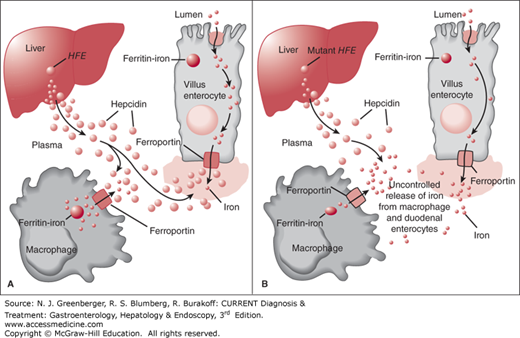
Investigators have shifted their focus from the duodenum to the liver, where the protein hepcidin is now considered the key regulator of iron absorption. It was first described by Park and colleagues, who named it after its site of synthesis in the liver and its antibacterial properties. Hepcidin is a 25 amino acid peptide encoded by the HAMP gene. There is an inverse relationship between the level of hepcidin and iron absorption. In mouse models of hereditary hemochromatosis, decreasing hepcidin production by silencing the HAMP gene encoding hepcidin results in severe iron overload. In iron-deficient mice, hepcidin production is also decreased, leading to increased iron absorption. In HFE knockout mice with iron overload, stimulating hepcidin production successfully reverses the iron overload.
Similarly, humans with HFE-related hereditary hemochromatosis have low levels of hepcidin mRNA in liver biopsy specimens despite iron overload. Weinstein and colleagues reported on two patients with large hepatic adenomas overexpressing hepcidin who presented with severe microcytic anemia. Resection of the masses reversed the hematologic abnormalities.
A recent study by Bordou-Jacquet and colleagues further supports a central role for the liver and hepcidin in iron regulation. They evaluated 18 C282Y homozygotes undergoing liver transplantation. Of the 18 patients, 11 had iron levels and hepcidin measurements available before and after liver transplantation. Before transplantation, all 11 had low hepcidin levels. After transplantation, the hepcidin level normalized in 10 of the 11, with a persistently low hepcidin level in one patient with iron deficiency. After transplantation, 9 of 11 had a persistently normal transferrin saturation without phlebotomy. Two patients had high iron levels, one with hereditary spherocytosis and the other with metabolic syndrome. Liver transplantation corrected the hepcidin dysregulation and iron overload associated with the HFE genotype, strongly supporting a central role of the liver and hepcidin in iron regulation.
However, in his editorial to this important study, Dr. Paul Adams notes that hepcidin does not appear to be the primary defect in hereditary hemochromatosis. Instead, a low hepcidin level appears to result from a cascade of events that starts with HFE mutations. Other newly discovered iron regulatory proteins, such as erythroferrone, can regulate hepcidin levels. In addition, comodifying genes such as TMPRSS6 likely contribute to the variable phenotypic expression of at risk HFE genotypes. Therefore, a low hepcidin level may be the final common pathway for a complex interplay of genetic and environmental events. Furthermore, a low hepcidin level may be necessary but not sufficient for the development of phenotypic hemochromatosis.
The pathways through which HFE mutations lead to iron overload are incompletely understood. One theory is that HFE is a participant in hepatocyte sensing of body iron status, perhaps as part of a hepatocyte sensing unit together with TfR1 and TfR2. Hepatocytes elaborate TfR2 receptors on their surface which may bind to diferric transferrin (Tf) in portal blood, possibly sensing the circulating level of iron by this means. HFE mutations could theoretically influence the binding and thereby the sensing of diferric Tf by hepatocytes and lead to lower levels of hepcidin. Another sensing unit may include hemojuvelin (HJV) together with two other proteins, BMP6 and neogenin (see non-HFE HHC below for more on HJV). These two sensing units may work independently or together to modulate hepcidin expression in the nucleus of the hepatocyte through a common intracellular signal transduction cascade. The intermediaries in this cascade likely include a bone morphogenetic protein (BMP) ligand on the cell surface and intracellular mediators and modulators known as SMAD proteins. A recent study indicates that HFE normally interacts with the BMP type 1 receptor ALK3 on the cell surface to regulate hepcidin expression. The HFE mutations C282Y and H63D may lead to decreased ALK3 on the cell surface which in turn downregulates hepcidin expression.
Hepcidin appears to have several targets. The primary target is ferroportin, the main iron exporter from mammalian cells such as duodenal enterocytes. When hepcidin binds to ferroportin at the basolateral cell surface of enterocytes, this leads to ferroportin internalization and degradation. This, in turn, limits iron export from enterocytes, leading to accumulation of iron within these cells and decreased iron absorption. Reticuloendothelial macrophages also have ferroportin receptors, and the effects of hepcidin here are thought to be similar.








