Disease
Complement Pattern
Lupus nephritis
Low C3 and C4
Peri-infectious (endocarditis, osteomyelitis, visceral abscess)
Low C3 and C4
Postinfectious (poststreptococcal)
Low C3; low (slight) or normal C4
Membranoproliferative GN
Low C3; normal C4
GN/RPGN are divided into three groups according to mechanism of disease (Table 10.2).
GOODPASTURE SYNDROME
Goodpasture syndrome is characterized by (1) proliferative crescentic GN, (2) pulmonary hemorrhage, and (3) anti-GBM antibodies. A renal-limited anti-GBM nephritis can also occur.
Pathogenesis
Circulating autoantibodies directed against an antigen (collagen IV epitope) intrinsic to the basement membranes in the glomeruli and alveoli result in tissue injury (Wieslander et al., 1984). It is possible that pulmonary manifestations occur in the setting of preexisting lung injury, whereas patients without previous lung injury present with renal-limited disease.
Causes of GN | |
|
■ Hematuria with RBC casts
■ Proteinuria
■ Anti-GBM antibodies
■ Chest x-ray—infiltrates corresponding to hemorrhage
■ Hypoxemia if severe
Pathology
■ Light microscopy
■ Focal and segmental endocapillary and extracapillary (crescents) proliferation (Fig 10.1).
■ Diffuse crescentic inflammation is the most common
■ There may be tubulointerstitial nephritis
■ Immunofluorescence
■ Intense, linear stain for IgG and C3 involving the glomerular basement membrane and sometimes also tubular basement membrane (Fig. 10.2).
■ Electron microscopy
■ No electron dense deposits (“pauci-immune”)
■ Widening of the subendothelial space with fibrin
■ Gaps in the glomerular basement membrane and Bowman capsule
Treatment
Plasmapheresis removes the circulating anti-GBM antibodies, whereas immunosuppressive therapy prevents new antibody formation and controls the inflammatory process. These treatments are very effective at treating pulmonary hemorrhage, but the kidneys may be less responsive.
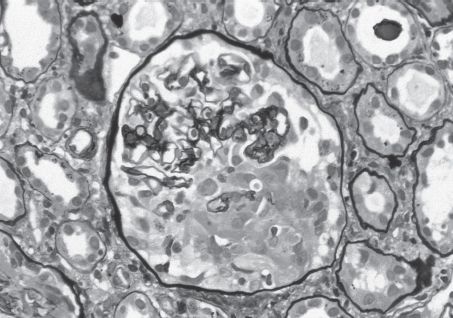
FIGURE 10.1 Anti-GBM nephritis. Early segmental fibrinoid necrosis with early cellular crescent formation. This light microscopic finding is that of a pauci-immune necrotizing GN, with a specific diagnosis of anti-GBM antibody-mediated GN made by immunofluorescence (Jones silver stain; original magnification ×400). (Originally published in Am J Kidney Dis 32(3): E1, 1998. Copyright by The National Kidney Foundation, with permission.) (See Color Plate.)
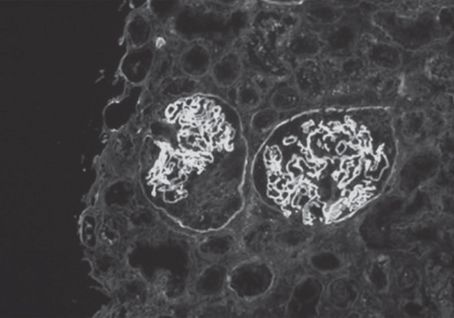
FIGURE 10.2 Anti-GBM nephritis. Linear staining of glomerular basement membranes with antibody to IgG and crescent formation in the glomerulus on the left, diagnostic findings of anti-GBM antibody-mediated GN (immunofluorescence with anti-IgG; original magnification ×200). (Originally published in Am J Kidney Dis 32(3): E1, 1998. Copyright by The National Kidney Foundation, with permission.) (See Color Plate.)
Systemic lupus erythematosus (SLE) is characterized by skin rash, oral ulcers, photosensitivity, myalgias, neurologic sequelae, arthritis, serositis, and renal involvement, the latter of which may undergo remissions and exacerbations both spontaneously and in response to treatment. Renal prognosis correlates with type and degree of glomerular involvement. Hence, pathologic classification guides treatment and predicts prognosis.
Pathogenesis
Circulating antibodies (including anti-DNA antibodies) formed to an unknown (possibly viral) antigen deposit in the kidney in locations dependent on the size and charge of the antigen and antibody (Hahn, 1998; Isenberg & Collins, 1985). Activation of complement and other inflammatory mechanisms lead to tissue injury.
Laboratory Evaluation
■ ANA panel (including anti-dsDNA and other autoantibodies)
■ Complements (C3, C4), CH50 (total hemolytic complement)—these decrease with disease exacerbation and normalize with disease improvement.
■ Hence, complements, ANA, anti-dsDNA, and urinalysis (hematuria, proteinuria) can be used to monitor disease progress. In selected cases, rebiopsy is needed
Pathology
■ Light microscopy reveals focal (<50% of glomeruli involved) or diffuse (>50% of glomeruli involved, often associated with crescents) involvement. Karyorrhexis (destructive fragmentation of the nucleus of dying cells) and “wire loop” thickening of basement membranes (due to immune deposits) are seen in severe disease. The WHO classification used for LN classification modified by the International Society of Nephrology/Renal Pathology Society (2003) is currently used
■ Class I—Minimal mesangial involvement
■ Class II—Mesangial proliferative LN
• Classes I and II are associated with a good prognosis and no therapy is necessary.
■ Class III—Focal proliferative LN (<50% of glomeruli involved)
• There is no consensus as to how Class III should be treated. Patients with only mild or moderate proliferative lesions involving a few glomeruli have a good prognosis and may respond to a short course of high-dose corticosteroids. Severe lesions with necrotizing features and crescents will require vigorous therapy (see Class IV).
■ Class IV—Diffuse proliferative LN (>50% of glomeruli involved, often associated with crescents) (Fig. 10.3)
• Requires aggressive treatment. Cyclophosphamide and steroids are standard treatment, though in some populations (African Americans, Asians), mycophenolate can be substituted for cyclophosphamide. Azathioprine, cyclosporine, rituximab (anti-CD20) are also used.
■ Class V—Membranous nephropathy
• May be indolent or progressive
• For patients with a good prognosis (subnephrotic proteinuria, preserved glomerular filtration rate [GFR]), cyclosporine plus corticosteroids may be appropriate. For patients with a poor prognosis (African Americans with nephrotic syndrome), treatment as for Class IV is appropriate
■ Class VI—Advanced sclerosing LN
• Greater than 90% of glomeruli are sclerosed. Immunosuppressive therapy is not indicated.
■ Immunofluorescense—Immune deposits to IgG, IgM, IgA, C3, C1q in all compartments (“full house”)
■ Electron microscopy—Electron dense granular deposits, tubuloreticular inclusions (intracytoplasmic tubular branching structures in cisternae of endoplasmic reticulum). Location of deposits in subendothelial location in Classes III and IV (Fig. 10.4) and in subepithelial location in Class V; in addition, mesangial deposits are virtually universal
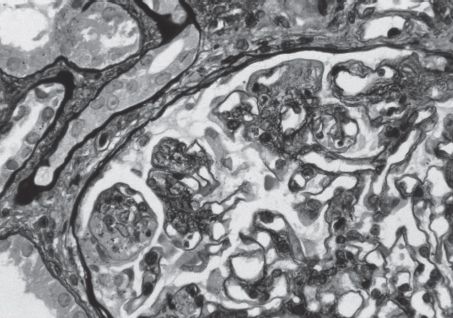
FIGURE 10.3 Lupus nephritis. Diffuse proliferative LN (WHO Class IV). There is segmental glomerular basement membrane splitting with eosinophilic, large, sausage-shaped subendothelial deposits. In other areas, holes in the basement membrane are seen, representing subepithelial deposits (deposits do not take up the silver stain). Segmental endocapillary proliferation and mesangial proliferation are also present (Jones’ silver stain; original magnification ×600). (Originally published in Am J Kidney Dis 31(6): E1, 1998. Copyright by The National Kidney Foundation, with permission.) (See Color Plate.)
Treatment
Treatment of LN is based on pathology (see above). In addition to or instead of immune-complex disease, some patients with SLE will have renal findings of antiphospholipid syndrome (renal microthombi) and are not treated with immunosuppressives but instead anticoagulation; if severe, plasmapheresis may be indicated.
IGA NEPHROPATHY
IgA nephropathy (nephritis) is characterized by IgA deposition in the mesangium with mesangial proliferation. Clinical features can range from asymptomatic hematuria to RPGN. Prognosis is usually good, but patients with abnormal renal function, severe proteinuria, or hypertension are at risk of renal failure. Though usually an idiopathic disease, it can be secondary to rheumatologic disease (especially ankylosing spondylitis) or liver disease (due to impaired clearance of IgA immune complexes from the portal circulation).
Pathogenesis
It is postulated that altered mucosal immunity in the setting of exposure to an environmental antigen leads to increased IgA synthesis. If there is abnormal galactosylation of the IgA, there will be decreased clearance by the liver, autoantibody formation to IgA1, and mesangial deposition (Mestecky et al., 1993).
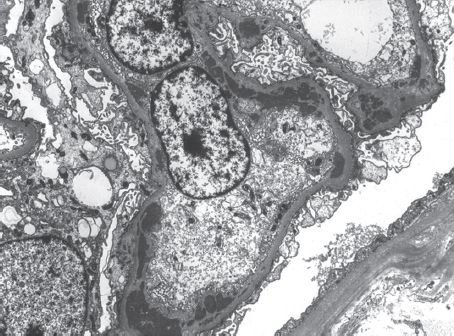
FIGURE 10.4 Lupus nephritis. Endocapillary proliferation and subendothelial deposits characteristic of proliferative LN are illustrated in this electron micrograph. Occasional small subepithelial, as well as mesangial, deposits are also present. The capillary loops are segmentally occluded by proliferating endothelial and mesangial cells (transmission electron micrograph; original magnification ×8,000). (Originally published in Am J Kidney Dis 31(6): E1, 1998. Copyright by The National Kidney Foundation, with permission.)
Laboratory Evaluation
■
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


