Fig. 5.1
Schematic of the adenoma-to-carcinoma sequence in the development of colorectal cancer. The primary genes that are mutated related to each step are indicated (See text for abbreviations and descriptions of the genes) (Adapted from Davies et al. [110]. With permission from Macmillan Publishers Ltd)
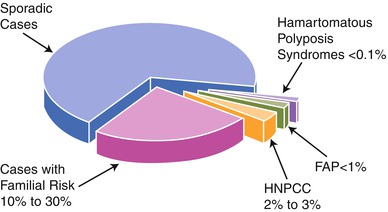
Fig. 5.2
Graph depicting the percentages of colorectal cancer patients that are considered to have sporadic, familial, and hereditary etiologies for their cancers (Reprinted from Burt [111]. With permission from Elsevier)
A third path for tumorigenesis is via DNA repair genes. These genes include mismatch repair (MMR) genes (e.g., MLH1, MSH2, etc.) as well as nucleotide- and base-excision repair genes (e.g., MYH). These genes are responsible for repair of DNA replication mistakes occurring during cellular division or those induced by exposure to environmental mutagens. If these genes are inactivated, DNA replication errors that routinely occur in every one in 1,000 DNA base pairings during cellular division are allowed to propagate through the daughter cell lines, with risks of subsequent alterations in other critical genes increasing (including proto-oncogenes or tumor suppression genes), leading to increased risk of cancer development [3]. Mutations in these types of genes can also be germline mutations, having been linked with inherited CRC syndromes such as hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch syndrome. More commonly than through germline mutations, carcinogenesis can occur through somatic mutations of these genes (as well as tumor suppressor genes), but gene inactivation can also occur through hypermethylation “silencing” of these genes’ promoter regions, much like through the serrated pathway, which is known as an epigenetic phenomenon since the DNA itself is not mutated.
Oncogenes
When proto-oncogenes, which regulate cellular proliferation and differentiation, become mutated, they may become oncogenes producing unregulated transcription or growth factors. This can occur through a number of mechanisms including missense mutations (point mutations leading to an amino acid change altering gene expression or protein function), chromosome rearrangement (altering gene expression or protein function), or through gene amplification (copy number increase of a portion of a chromosome leading to increase in gene expression). Oncogenes behave such that a mutation in one of the two alleles is sufficient to produce activation and phenotypic expression of the mutated gene. Oncogenes implicated in sporadic CRC include RAS genes, c-Myc, and BRAF.
RAS Family
The RAS oncogene family includes three cellular variants Harvey- RAS (HRAS), Kirsten-RAS (KRAS) and Neuroblastoma-RAS (NRAS). KRAS is the most commonly mutated gene in CRC from the RAS family and lies on the short arm of chromosome 12 (12p), encoding for a guanosine triphosphatase (GTPase) protein involved in the transduction of growth and differentiation signals through the serine protein BRAF (see below) [6]. When mutated and activated, KRAS results in cellular overgrowth and dysplasia, likely as an early event in tumorigenesis, usually found after the initial APC mutation in CRC development (Fig. 5.1). KRAS mutations are present in about 50 % of CRC and colonic adenomas ≥1 cm compared with only 9 % of adenomas <1 cm, suggesting that in a proportion of CRCs, RAS activation is an early promoter rather than an initiator of tumorigenesis [7]. KRAS has also been implicated in the process of tumor invasion and metastasis. A study conducted comparing genetic and epigenetic changes in primary metastatic and non-metastatic CRC found that KRAS mutations were significantly associated with metastatic tumors [8].
Importantly, if particular KRAS mutations are present in CRC, there appears to be an impact on the targeted response of epidermal growth factor receptor (EGFR) agents such as cetuximab and panitumumab [9]. These monoclonal antibodies to EGFR are thought to work through binding of the agents leading to internalization of the receptor and blockage of downstream KRAS signalling. It is believed that KRAS mutations leading to a constitutively active protein (>90 % of the KRAS mutations, located on codons 12 and 13 in exon 2 of the gene) will negate the effects of the EGFR agent [10]. This has been confirmed in clinical studies. Karapetis, et al. analyzed 394 out of 572 patients with CRC who were randomly assigned to receive cetuximab plus best supportive care or best supportive care alone with the effectiveness of cetuximab being significantly associated with KRAS mutation status (P = 0.01). Patients with wild-type KRAS tumors showed significantly improved survival (median survival: 9.5 vs 4.8 months) with treatment with cetuximab compared with supportive care alone whereas patients with a mutated KRAS showed no difference to those who received supportive care alone with respect to overall survival or progression free survival [9]. This finding has been confirmed by meta-analyses of multiple studies on EGFR agents and KRAS status [11, 12] as well as recent trials [13, 14]. A number of European and North American Oncology Societies have recommended that all patients with metastatic CRC who are being considered for anti-EGFR therapy have their cancer tested in an accredited lab for KRAS mutations as standard of care [10].
NRAS, a membrane protein very homologous with KRAS, is found on chromosome 1 and is mutated in approximately 3–5 % of CRCs [10]. Mutations in this gene are often mutually exclusive from those in KRAS [15]. In a European Consortium trial with cetuximab, in KRAS non-mutated patients, NRAS mutants had a significantly lower response rate (7.7 % vs. 38.1 %; OR, 0.14; p = 0.013) than did NRAS wild types, and a trend for shorter progression free and overall survival [10]. Similarly, a 2013 study reviewing outcomes from 1,060 patients who had both KRAS and NRAS testing of their tumor showed worse outcomes (response rate and survival) with the use of panitumumab in the setting of any RAS mutation versus wild type tumors with the same chemotherapeutic regimen. The authors of this study concluded that anti-EGFR agents “…have no value in patients with metastatic colorectal cancer and mutated RAS” [14]. The results of these studies have impacted treatment guidelines such that many organizations advocate KRAS and NRAS (and possibly BRAF, if needed) testing of all metastatic tumors due to the impact of the results on treatment regimens [16].
c-Myc
The proto-oncogene c-Myc located on chromosome 8 has been associated with a number of cell functions including production of a transcription factor linked with cellular functions such as differentiation and apoptosis as well as tumor angiogenesis [17]. While the gene is most frequently linked with Burkitt’s lymphoma, it is clearly involved in CRC tumorigenesis and has shown overexpression in the majority of CRCs. While it has not been used to direct therapeutics like the RAS genes have, c-Myc overexpressing CRCs have shown better survival, although this advantage appears to be negated in the presence of a mutant p53 gene [18].
BRAF
BRAF is a signal transduction gene on chromosome 7q34 involved in the MAP kinase cellular pathways (associated with RAS) that impact cell division, proliferation, and apoptosis. BRAF is mutated in 5 % of sporadic “adenoma to carcinoma” CRCs but up to 25 % of all CRCs (Fig. 5.3). Approximately 90 % of the time the BRAF mutation is in the form of a substitution at nucleotide 1799 leading to valine (V) being substituted for by glutamate (E) at codon 600 (referred to as “V600E”) which leads to a ten times more active BRAF protein than normal [19]. As noted above, BRAF mutations are often mutually exclusive of KRAS and NRAS mutations, and BRAF is less frequently found mutated when a CRC is associated with p53 and APC mutations, suggesting that BRAF mutations may be part of a different CRC tumorigenesis pathway [1]. This pathway is often thought to be related to epigenetic silencing of MLH1 due to hypermethylation of its promoter region leading to a sporadic microsatellite instability high (MSI-H, see below) CRC developing via the CpG island methylator phenotype (CIMP) mechanism also known as the “serrated pathway” described by Jass. In fact, the odds ratio of an association between CIMP and a BRAF mutation in CRC is greater than 200 [20]. CRCs associated with BRAF mutations and the CIMP-high (CIMP-H) pathway are more often in older women, right side of the colon, and associated with smoking [21]. BRAF mutations are found in 40–87 % of sporadic MSI-H CRCs [22], but the order of molecular events leading to MSI-H and/or CIMP-H CRCs has not been completely defined when associated with BRAF mutations [21]. When a BRAF mutation is noted in association with an MSI-H CRC, the tumor is more likely to be a sporadic CIMP-H CRC as opposed to related to the hereditary condition Lynch syndrome caused by a germline MMR gene mutation, and therefore BRAF mutation testing can be used to determine the need for further MMR germline testing [19, 23]. BRAF testing is also indicated in CRCs that are KRAS and NRAS wild type if the patient is being considered for anti-EGFR therapy, as discussed above. Finally, the presence of a BRAF mutation in the setting of a microsatellite stable CRC (as opposed to MSI-H) is a poor prognostic indicator, although this does not yet inform therapy recommendations at the present time [19].
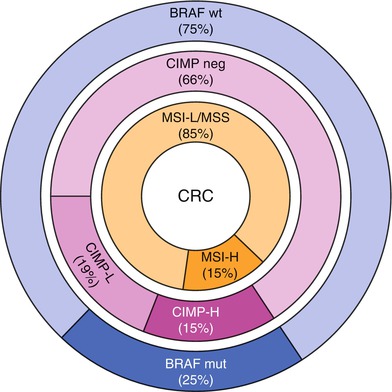
Fig. 5.3
Molecular etiologies of colorectal cancer which can be characterized by the presence or absence of microsatellite instability (MSI), the CpG island methylator phenotype (CIMP), or BRAF mutation status (wild type [wt] or mutant [mut]). MSI and CIMP are characterized as “high” (MSI-H and CIMP-H, respectively) and “low” (MSI-L and CIMP-L, respectively) (From Boland and Goel [112]. With permission from Oxford University Press)
Tumor Suppressor Genes
This class of genes inhibits abnormal cell growth by slowing the cell cycle to allow for DNA repair and promote apoptosis when repair is no longer possible. They are recessive genes, meaning that both alleles must be lost or mutated for the gene to be inactive and phenotypically expressed. This inactivation can occur through a number of mechanisms including deletion or mutations that impact protein structure or function (nonsense mutations that lead to a truncated, nonfunctional protein, frameshift mutations that lead to translation of a different protein, or splice site mutations which leave introns present in the RNA that then translate an incorrect protein product). Tumor suppressor genes that play a role in CRC include APC, DCC, and p53.
Adenomatous Polyposis Coli (APC)
The APC gene is located on the long arm of chromosome 5 (5q) [24]. It encodes a protein that has multiple functional domains that mediate oligomerization as well as binding of a variety of intracellular proteins including β-catenin, ϒ-catenin, glycogen synthase kinase (GSK)-3β, Axin, tubulin, EB1 and hDLG [25]. Mutant, truncated APC proteins lack at least one type of β-catenin binding repeat. The APC protein is located in the basolateral membrane in the colorectal epithelial cells, and as the cells migrate up through the crypt columns, expression increases [26]. In colorectal mucosal cells, damage to the APC protein complex results in increased levels of free cellular β-catenin. Two important functions of β-catenin include cellular adhesion and cell signaling. Therefore, with a dysfunctional truncated protein related to the APC gene mutation, whether sporadic or germline, β-catenin accumulates in the nucleus and induces gene overexpression through aberrant cell signalling (via the Wingless [Wg] and Wnt signaling pathways first described in Drosphilia and mice) and increased cell proliferation. Additionally, since binding of β-catenin to cadherins (important for cell-to-cell adhesion) and to the APC protein is mutually exclusive [25], the cytoplasmic accumulation of β-catenin due to the presence of a dysfunctional non-binding APC protein enhances cell-to-cell adhesion and limits cell migration. This results in the balance of cellular turnover shifting from the lower proliferative compartment of the crypt to the upper villi, which is greatly impaired, leading to the accumulation of the hyperproliferating cells [27]. In addition, the truncated APC protein acts through the Wnt/β-catenin/Axin signalling pathway to alter apoptosis and cell cycle control which drives neoplastic cell proliferation further leading to the formation of an adenomatous, pre-cancerous polyp [4].
Germline mutations in APC result in FAP or one of its variants such as attenuated FAP (aFAP) or Turcot syndrome [28–32]. “Classic” FAP is an autosomal dominant disease that is associated with <1 % of all incident CRCs and leads to carpeting of the colon and rectum with hundreds to thousands of adenomatous polyps. When germline mutations occur in the APC gene, there is a very high phenotypic penetrance with 90–100 % of mutation carriers ultimately developing FAP. An individual with FAP has one mutated copy of the APC gene, and a subsequent somatic inactivation of the second wild type copy of the gene in a colonic epithelial cell leads to polyp initiation as discussed above [33]. In “classic” FAP, polyps usually start developing in adolescence, and if the colon is left intact, CRC will develop at an average age of 39 years [34–36]. In contrast, aFAP (also linked with the MYH base-excision repair gene and presenting as an autosomal recessive condition) is defined by fewer than 100 colorectal adenomas with polyp onset usually in the mid-30s and CRC developing on average by the mid-50s. Varied clinical presentations can be dependent on the location of the mutation within the APC gene [37]. Mutations that are proximal to codon 1249 or distal to codon 1465 (at the 3′ and 5′ ends of the APC gene) usually lead to the aFAP phenotype, whereas mutations between codons 1250 and 1330 (especially 1309 and 1328, which have been associated with higher rates of rectal cancer) lead to more extensive polyposis.
Extracolonic malignancies also occur in FAP including hepatoblastoma in children, papillary thyroid cancer, duodenal and gastric carcinomas, and ampullary and pancreatic carcinomas, as well as benign disorders such as dermoid cysts, desmoid tumors, osteomas, supernumerary teeth, and congenital hypertrophy of the retinal pigment epithelium (CHRPE). These extracolonic manifestations are often also related to the APC mutation site (Fig. 5.4). For example, CHRPE is found primarily in patients with mutations located between exons 9 and 15. Turcot syndrome is associated with FAP and medulloblastoma of the central nervous system when related to an APC gene mutation, but it has also been associated with Lynch syndrome (the hereditary condition related to germline MMR gene mutations) [34–36].
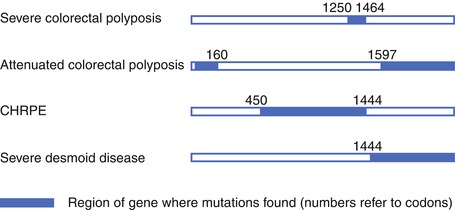
Fig. 5.4
Schematic of the APC gene to show some of the genotype-phenotype correlations noted in familial adenomatous polyposis (FAP) (From Beck et al. [113]. With kind permission from Springer Science + Business Media, Figure 37.3)
Genetic testing for FAP should first involve genetic counseling to ensure proper assessment for determination of the appropriateness of testing and to ensure that the patients and their families understand the implications of testing. Approximately 25–30 % of FAP patients will be related to de novo germline mutations, meaning that the parents of the affected patient do not carry the gene mutation or manifest the disease phenotypically. Mutation detection during testing depends on the phenotypic manifestations of FAP. In a study of over 7,000 patients with varying degrees of adenomatous polyposis, gene testing for APC and MYH were conducted with 80 % of those with “classic” FAP (>1,000 polyps) showing a pathogenic APC mutation and 2 % showing a biallelic MYH mutation. In contrast, those patients with between 20 and 99 adenomas had an APC mutation only 10 % of the time and a biallelic MYH mutation 7 % of the time [38]. Genetic testing other members of the family can be facilitated if the initially diagnosed affected proband has an abnormal test, allowing for mutation-specific testing of those at-risk as opposed to need for full gene sequencing [39]. Having abnormal genetic testing or being at high risk without undergoing genetic testing (e.g., child of an FAP patient) will require a lifetime of surveillance beginning with a flexible sigmoidoscopy or colonoscopy at age 10–15 years and then every 12 months thereafter, as well as an upper endoscopy starting as age 20 years (or before colectomy) and occurring every 1–4 years to assess for gastric, periampullary and duodenal adenomas [40, 41]. For patients who have or may have aFAP, surveillance should consist of a full colonoscopy starting in the late teens and every 2–3 years thereafter secondary to the predominance of right sided colonic tumors, and upper endoscopy is also advised starting at age 25–30 years [41]. The National Comprehensive Cancer Network has advised that all of these “…patients be managed by physicians or centers with expertise in FAP” [41].
Because of the CRC risk related to these conditions, total abdominal colectomy and ileorectal anastomosis (IRA) or total proctocolectomy (TPC) and either end ileostomy or restoration of intestinal continuity through an ileal pouch anal anastomosis (IPAA) is advised for patients with FAP and aFAP. The timing of the operation is dependent on the phenotypic manifestations (e.g., development of CRC or high grade dysplasia, etc.) as well as symptoms of the disease (e.g., bleeding) as well as the maturity and understanding of the patient, but the ideal time is thought to be during the late teens or early 20s.
Operative choice for FAP or aFAP is based on the patient’s phenotype (e.g., presence of cancer, rectal polyp number greater than 20, etc.) as well as their baseline health and continence status. Concerns about desmoid development (higher in women and after operations, especially in those with a family history of desmoids) and sexual function and fecundity (ability to become pregnant) may impact the decision of the operative approach as well. Concerns about rectal cancer and rectal polyp development impact operative choice as well. A Dutch Polyposis Registry study tried to assess the appropriate operative choice for FAP patients based on genotype by tracking those patients having undergone an IRA who were grouped based on the site of their APC gene mutation as an indicator of their polyposis phenotype. Rectal cancer risks ranged from 3 to 8 % in the rectal remnant and need for proctectomy during 20 years of follow-up ranged from 10 to 74 % depending on the initial phenotype, suggesting that more aggressive genotypes would benefit from TPC at the time of their initial operation [42]. An IPAA does not completely eliminate the risk of distal polyps or cancer issues, unfortunately, and therefore FAP patients that undergo IPAA require continued rectal remnant and pouch surveillance. A French group assessed adenoma development after IPAA for FAP with the risk of developing adenomas at 5, 10, and 15 years being 7, 35 and 75 %, respectively. No invasive carcinomas were noted. Interestingly, they did not find a correlation between adenoma development and the site of the APC mutation [43]. Also, a Dutch study on 254 patients after IPAA for FAP showed the cumulative risk of developing an adenoma in the pouch at 10 years to be 45 % with 12 % developing an advanced adenoma and 2 % developing carcinoma [44]. Therefore, depending on the polyp burden, pouchoscopy is recommended every 1–3 years after IPAA and proctoscopy every 6–12 months after IRA [41]. Chemoprevention options for FAP are discussed below.
p53
The p53 gene is located on the short arm of chromosome 17p [45]. It functions as the gatekeeper by slowing the cell cycle to allow for DNA repair after damage by ultraviolet light, radiation or chemotherapy [6]. Inactivation of p53, found in 70 % of CRC cases and as many as half of all carcinomas in humans, occurs late in the tumorigenic sequence. Therefore, p53 gene mutation is likely to be a limiting factor for the malignant transformation of precancerous cells in the LOH pathway. If a p53 mutation is identified in CRC it has potentially both prognostic and therapeutic significance. Most studies show a significantly lower survival rate for patients with p53 negative tumors compared with those with non mutated (wild type) p53 [46].
Deleted in Colon Cancer (DCC)
The DCC gene is located on the long arm of chromosome 18 (18q) [47] and codes for a transmembrane protein involved in cell-to-cell adhesion. It has been hypothesized that inactivation of the DCC gene and the resulting absence of the DCC protein may enhance the metastatic potential of CRC. DCC protein expression may also have prognostic significance and DCC-positive Stage II and III CRC were found to have statistically significant overall survival when compared with those with DCC negative cancers [48]. Studies are still ongoing as to the prognostic significance of DCC in CRC.
Mismatch Repair
The MMR genes are responsible for correcting incorrect nucleotide base pairings or small nucleotide insertions or deletions that occur routinely during DNA replication [49, 50]. These genes include MSH2 (mutS homolog 2), MSH6 (human mutS homolog 6), MLH1(mutL homolog 1), and PMS2 (post meiotic segregation 2), amongst others. When a patient has one mutated MMR gene, the normal allele is able to produce the proteins needed for DNA repair functions; but when the normal remaining gene undergoes a “second hit” mutation, the MMR proteins fail to form or function appropriately. If MMR errors subsequently occur in the replication of an oncogene or tumor suppressor gene, neoplasia may result.
Cancers due to a MMR gene mutation are associated with an MSI-H phenotype. Microsatellites are short, tandemly repeated DNA base sequences that are scattered throughout the genome, some being located near significant protein-encoding genes. When there is lack of one of the functional MMR proteins, there can be variability in these repeats which leads to MSI [51]. MSI is highly sensitive for Lynch syndrome, the hereditary CRC syndrome associated with an MMR germline mutation. Almost 90 % of CRCs from patients with Lynch syndrome are MSI-H while MSI is found in up to 15 % of sporadic CRCs [52, 53]. MSI-H CRCs, whether sporadic or related to Lynch syndrome, tend to occur in the proximal colon, have a greater mucinous component, contain lymphocytic infiltration, and are more often poorly differentiated [54]. These criteria were incorporated into the Bethesda and revised-Bethesda criteria (Table 5.1) which have been utilized to determine which CRCs warrant testing for MSI to then determine whether further genetic testing for Lynch syndrome might be warranted (see below).
Table 5.1
Revised Bethesda criteria for testing colorectal cancer for microsatellite instability (MSI)
Patients who meet Amsterdam criteria (Table 5.2) |
Colorectal cancer diagnosed in a patient below age 50 years |
Presence of synchronous and/or metachronous colorectal or other HNPCC-associated tumors (endometrial, stomach, small bowel, ovarian, pancreas, ureter and renal pelvis, biliary tract, and brain (usually glioblastoma) tumors, sebaceous gland adenomas and keratoacanthomas, and carcinoma of the small bowel), regardless of patient age |
Colorectal cancer with “MSI histology” (tumor infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous/signet-ring differentiation, or medullary growth pattern) diagnosed in a patient who is less than 60 years of age |
The condition often considered synonymous with Lynch syndrome is known as “HNPCC” or hereditary nonpolyposis colorectal cancer, which describes patients who fit clinical criteria that were originally developed to identify high risk patients for research studies (Amsterdam I and II criteria) (Table 5.2). Both HNPCC and Lynch syndrome are autosomal dominant disorders that are caused by germline mutations in the MMR genes. They account for 3–6 % of all incident CRCs, and patients with the condition are also at increased risk for extracolonic malignancies such as endometrial, genitourinary, central nervous system, biliary, and ovarian cancers, amongst others [55]. These patients often have early onset CRC (average age 42 years) with a high risk of synchronous (5–20 %) and metachronous (10–50 %) CRC [56]. Mutations in MLH1 are implicated in 32 % of Lynch syndrome cases, MSH2 mutations in 39 % of cases, MSH6 in 15 % of cases and PMS2 mutations in 14 % of cases [57]. The estimated penetrance of MMR gene mutations can be high, with anywhere from 30 to 80 % of MMR gene mutation carriers developing CRC and 20–60 % developing endometrial cancer. As in FAP, genotype-phenotype correlations have also been determined for Lynch syndrome. For example, Weber, et al. demonstrated that kindreds with MLH1 mutations had a higher incidence of rectal cancer and fewer extracolonic manifestations when compared with those with MSH2 mutations [54].
Table 5.2
Amsterdam II criteria for hereditary nonpolypoisis colorectal cancer (HNPCC)
≥3 relatives with an associated cancer (colorectal cancer, or cancer of the endometrium, small intestine, ureter or renal pelvis), one should be a first-degree relative of the other two |
≥2 successive generations affected |
≥1 relative diagnosed before age 50 years |
FAP has been ruled out |
Individuals with a germline MMR gene mutation or family history fulfilling HNPCC criteria should have a colonoscopy every 1–2 years starting between the ages 20 and 25 years and every year after age 40 years. A prospective trial screening 22 Lynch syndrome families demonstrated a significant 63 % decrease in the development of CRC from 11.9 to 4.5 % in those who underwent surveillance colonoscopy or barium enema plus sigmoidoscopy every 3 years compared to those who underwent no screening [58]. Studies have shown that appropriate screening of Lynch syndrome patients improves mortality rates as well [59]. The timing of screening initiation may be adjusted depending on the underlying genotype, especially with MSH6 and PSM2 mutation patients developing CRC later in life on average versus MLH1 and MSH2 patients [41].
Surgical options for colon cancer in the setting of Lynch syndrome include a segmental colectomy versus total abdominal colectomy with IRA (due to the risk of metachronous cancer). Three separate studies (two from single institutions and one from the multi-institutional Colon Cancer Family Registry (CCFR)) showed that more extended colon resections to treat CRC in HNPCC or Lynch syndrome patients lead to significantly decreased metachronous cancer rates, even when compliance was appropriate with post-operative screening protocols [60–62]. This has also been found when assessing the treatment of patients with Lynch syndrome and rectal cancer, which is the presenting index cancer in approximately 15 % of these patients [63]. One single institution study of HNPCC patients who initially presented with rectal cancer showed a greater than 50 % rate of metachronous advanced adenomas or colon cancers at a median of 6 years after proctectomy [64]. A CCFR study of 79 Lynch syndrome patients with an index rectal cancer who underwent proctectomy showed risks of metachronous colon cancer of 19 % at 10 years, 47 % at 20 years, and 69 % at 30 years [65]. Authors of both studies concluded that HNPCC/Lynch syndrome patients should be considered for TPC and IPAA at the time of index rectal cancer treatment.
Similar to FAP, the genetic testing for Lynch syndrome must start with genetic counseling and familial assessment. There are a number of algorithms utilized to determine which patients with CRC should be tested for Lynch syndrome. The Bethesda Guidelines (Table 5.1) were established in 1996 to help identify which CRC patients to consider for further Lynch syndrome testing via MSI testing [66]. Because of the inaccuracy of family histories and studies showing a close to 50 % miss rate for Lynch syndrome using clinical criteria alone [67], universal screening of all CRCs was recommended by some national organizations, whether through MSI testing or use of immunohistochemistry assessment of tumor MMR proteins [68]. Attempting to identify the best Lynch syndrome screening strategy for CRCs, one study of over 10,000 patients assessed the sensitivity and specificity of universal screening (sensitivity, 100 %; specificity, 93.0 %) versus Bethesda guidelines (sensitivity, 87.8 %; specificity, 97.5 %). Meanwhile, a strategy known as Jerusalem criteria (screen all CRC patients 70 years old or younger) showed sensitivity of 85.4 % and specificity of 96.7 % while a strategy based on Jerusalem criteria as well as screening those over 70 years fulfilling the Bethesda guidelines showed sensitivity of 95.1 % and specificity of 95.5 % with the latter option missing almost 5 % of Lynch syndrome cases but resulting in 35 % fewer cases undergoing unnecessary MMR testing [69]. Regardless of strategy, institutions are recommended to identify a means to screen CRCs for Lynch syndrome. As mentioned above, BRAF or hypermethylation testing should also be considered as part of the screening strategy to rule out an epigenetic CRC etiology [23].
Colorectal Cancer Screening
Since the introduction of nationwide CRC screening efforts in the United States, the death rate related to the disease has been slowly declining. Since 1998, CRC-related mortality rates have decreased by 3 % per year in men and by 2.3 % per year in women (American Cancer Society). This has been attributed to a number of factors, including the effect of screening identifying more early, treatable CRCs as well as even preventing the initial development of CRC through removal of adenomas endoscopically. This reduction is likely also related to reduced exposure to CRC risk factors in the general population as well as improving treatment modalities for CRC and rectal cancer alike [70].
Unlike other screening programs which aim to just identify cancers early enough in their development such that they are curable (e.g. mammography for breast cancer, prostate specific antigen screening for prostate cancer, etc.), screening for CRC can both find early, treatable CRCs as well as prevent these cancers from developing in the first place through removal of their adenoma precursors [71]. These precursors especially include those polyps at highest risk for malignant degeneration including advanced adenomas (adenomas ≥10 mm in size, those with high grade dysplasia, and/or those with a villous component) [72]. Because screening the entire population for CRC is not possible or cost-effective, the available screening modalities are recommended based on their efficacy as well as stratifying patients into average, intermediate or moderate risk, and high-risk categories. These designations help determine the best modality of screening as well as the age of initiation of screening and the appropriate screening intervals.
Those who are considered average risk for CRC, which includes 70–80 % of all patients eligible for screening, are those who are 50 years and older, asymptomatic, and without other risk factors such as family or personal history of colorectal neoplasia. Moderate/intermediate risk accounts for 15–20 % of the eligible screening population and includes those with a family history of CRC or adenomas diagnosed at age < 60 years in one or more first degree relatives (parent, sibling or child) or two first degree relatives at any age. Personal history of CRC or adenomatous polyps also puts patients in a moderate risk category. High risk (5–10 % of all those eligible for CRC screening) includes those with a known family history of FAP, HNPCC/Lynch syndrome or other inherited CRC syndrome or a personal history of inflammatory bowel disease (IBD) including Crohn’s colitis or ulcerative colitis. Most of the recommendations below are from The Colorectal Cancer Screening and Surveillance recommendations of the U.S. Multisociety Task Force on Colorectal Cancer [73]. Tables 5.3 and 5.4 show the summary recommendations.
Table 5.3
Guidelines for screening for the early detection of colorectal cancer and adenomas for average‐risk women and men aged 50 years and older
The following options are acceptable choices for colorectal cancer screening in average adults beginning at age 50 years, Since each of the following tests has Inherent characteristics related to prevent on potential, accuracy, costs, and potential harms, individuals should have an opportunity to make an informed decision when choosing one of the following options | ||
In the opinion of the guidelines development committee, colon cancer prevention should bе the primary goal of colorectal cancer screening. Tests that are designed to detect both early cancer and adenomatous polyps should be encouraged if resources are available and patients are willing to undergo an invasive test | ||
Test | Interval | Key issues for informed decisions |
Tests that detect adenomatous polyps and cancer | ||
FSIG with insertion in 40 cm or to splenic flexure | Every 5 years | Complete or partial bowel prep is required |
Sedation usually is not used, so there may be some discomfort during the procedure | ||
The protective effect of sigmoidoscopy is primarily limited to the portion of the colon examined | ||
Patients should understand that positive findings on sigmoidoscopy usually result in a referral for colonoscopy | ||
Colonoscopy | Every 10 years | Complete bowel prep is required |
Conscious sedation is used in most centers; patients will miss a day of work and will need a chaperone for transportation from the facility | ||
Risks include perforation and bleeding, which are rare but potentially serious; most of the risk is associated with polypectomy | ||
DCBE | Every 5 years | Complete bowel prep is required |
If patients have one or more polyps ≥6 mm, colonoscopy will be recommended; follow-up colonoscopy will require complete bowel prep | ||
Risks of DCBE are low, rare cases of perforation have been reported | ||
CTC | Every 5 years | Complete bowel prep is required |
If patients have one or more polyps ≥6 mm, colonoscopy will be recommenced; if same day colonoscopy It not available, a second complete bowel prep will be required before colonoscopy | ||
Risks of CTC are low; rare cases of perforation have been reported | ||
Extracolonic abnormalities may be identified on CTC that could require further evaluation | ||
Tests that primarily detect cancer | ||
gFOBT with high sensitively for cancer | Annual | Depending on manufacturer’s recommendations, two to three stool samples collected at home are needed to complete testing, a single sample of stool gathered during a digital exam in the clinical settling is not an acceptable stool test and should not be done
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




