, Mark Thomas1 and David Milford2
(1)
Department of Renal Medicine, Birmingham Heartlands Hospital, Birmingham, UK
(2)
Birmingham Children’s Hospital, Birmingham, UK
Abstract
In this chapter we explain:
Anaemia due to kidney disease
Kidney disease linked to causes of anaemia
The importance of acidosis in chronic kidney disease
Effects of kidney failure on bone and mineral metabolism
Hyperparathyroidism and the effects of parathyroidectomy
How hypercalcaemia is linked to kidney disease
As kidney function declines, three main laboratory test abnormalities develop (Fig. 13.1).
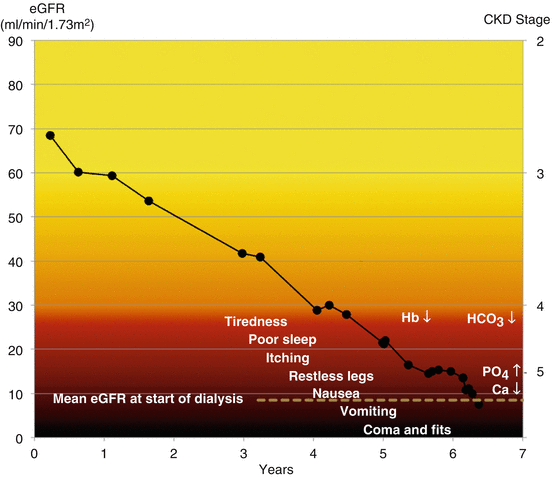
Fig. 13.1
Laboratory test abnormalities and symptoms that occur at different stages of chronic kidney disease
Low Hb – anaemia
Low serum bicarbonate – acidosis
Low calcium and raised phosphate and PTH – mineral bone disease
Anaemia
Erythropoietin and the Regulation of Haemoglobin
Anaemia in kidney disease is due to insufficient production of erythropoietin combined with a shortening of the life span of red blood cells.
Erythropoietin increases the production of red cells by binding to specific receptors on erythroid progenitor cells in the bone marrow, triggering a cascade of intracellular protein phosphorylations. As a consequence of this activation, the cells proceed to form mature red cells rather than undergo apoptosis.
In the fetus and newborn babies, erythropoietin is produced predominantly by the liver. In infancy production shifts to the kidneys as their blood flow and function increases. In adults, 90 % of erythropoietin comes from the kidneys.
Erythropoietin production is controlled by changes in the pressure of oxygen (pO2) dissolved in tissue fluid. The oxygen sensing takes place inside interstitial fibroblast cells , located between the tubules in the deep layers of the renal cortex close to the peritubular capillaries [1].
The control of erythropoietin production is mediated by hypoxia-inducible factors (HIFs), which promote transcription of the erythropoietin gene. When pO2 is normal, HIF is rapidly hydroxylated by oxygen-dependent HIF prolyl-4-hydroxylase domain (PHD) enzymes and so does not accumulate in the interstitial cell. Under hypoxic conditions the PHD enzymes are not activated; HIF levels increase in the cell and turn on the erythropoietin gene. Erythropoietin levels in blood may increase 1000-fold in anaemia not related to CKD.
As GFR drops, the haemoglobin concentration falls (Patient 13.1). The reduction in erythropoietin production in kidney disease is due to reduced responsiveness of the interstitial cells to hypoxia from reduced HIF activity, combined with a reduced capacity to produce erythropoietin. If HIF levels are increased by pharmacological inhibition of the PHD enzymes, erythropoietin production increases [2].
Patient 13.1: The Development of Anaemia in Chronic Kidney Disease
Mr. Roberts was under regular follow up for CKD and hypertension. Over the course of a year his eGFR dropped further, by half. An ultrasound scan showed a shrunken right kidney.
As the right kidney atrophied, glomerular filtration and erythropoetin production declined, leading to a decline in eGFR and haemoglobin concentration (Fig. 13.2).
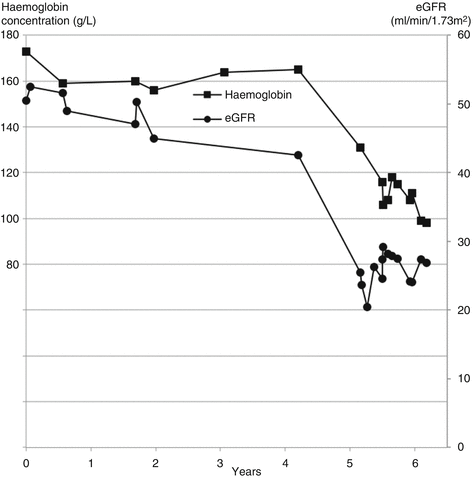
Fig. 13.2
Parallel reductions in eGFR and haemoglobin concentration due to atrophy of one kidney in a patient with pre-existing chronic kidney disease
Why in the Kidneys?
Why are the kidneys central to the regulation of red cell production by the bone marrow? The answer to this question becomes clear from thinking about the role of haemoglobin in survival and evolution.
The optimal haemoglobin (Hb) concentration is the one that delivers oxygen most efficiently to the tissues. Too high a concentration increases the risk of hyperviscosity and clotting and too low risks high output cardiac failure. The regulatory mechanism maintains a basal rate of red cell production to replace red cells that are cleared from the circulation at the end of their life. When the Hb concentration falls below the optimum level, the regulatory mechanism stimulates red cell production.
A low Hb concentration provides an inadequate supply of O2 to the tissues. This may be compensated by an increase in blood flow, for example in exercising muscles by up to 20-fold. Tissues that are unable to compensate for anaemia by increasing their blood flow are vulnerable to hypoxic damage.
Kidney blood flow has to be closely regulated. If the blood flow rate increased in response to anaemia, GFR would also increase. This would increase the amount of O2 and energy required by the tubules to reabsorb the increased amount of filtered sodium. The increased O2 consumption would negate the extra O2 provided by the increased blood flow.
Because hypoxia cannot be compensated by increased blood flow, the kidneys are at risk of hypoxic damage due to anaemia. Locating the haemoglobin regulatory mechanism in the region of the kidney most at risk – the deep cortex – provides the greatest survival advantage.
The anaemia of CKD provides some survival advantage against cardiovascular disease. Blood clotting is impaired due to anaemia and impaired platelet function. The risk of stroke increases if the anaemia is corrected with recombinant erythropoietin [3].
Anaemia has also indirectly increased survival from renal failure through dialysis. The success of haemodialysis is possible because of the arterio-venous fistula that allows repeated safe access to the circulation (see Sect. “Vascular access” in page 255). If one attempts to create a fistula in someone with normal kidney function, the vein clots almost immediately. Similarly, if a dialysis patient regains normal kidney function with a transplant, the fistula usually clots. The anaemia and bleeding tendency of renal failure have permitted the evolution of haemodialysis by allowing arterio-venous fistulas to function.
The red cells are normal in CKD – the anaemia is normochromic normocytic – and the other cells lines are normal. If someone with CKD has abnormal red cells or changes in white cells or platelets, an alternative cause for the anaemia should be sought. Common causes include iron deficiency from gastrointestinal blood loss or coeliac disease and folate or vitamin B12 deficiency causing macrocytic anaemia.
Kidney Disease Linked to Causes of Anaemia
The cause of the anaemia may sometimes also be the cause of the kidney disease. Sickle cell anaemia is common; paroxysmal nocturnal haemoglobinuria (PNH) and haemolytic uraemic syndrome (HUS) are rare. In all three, anaemia is caused by intravascular haemolysis. If haemolysis is suspected, check for a raised serum lactate dehydrogenase (LDH) , which is released from the red cells.
Sickle cell anaemia can cause infarction of the renal medulla and papillae through sickling of red cells in this hypoxic region. Necrotic papillae slough off into the pelvis and can cause haematuria, ureteric colic and obstruction.
Chronic medullary ischaemia causes haematuria and impairs the kidney’s ability to concentrate urine and excrete acid and potassium. Patients may have a wide range of clinical presentations including haematuria, proteinuria, nephrotic syndrome , acute and chronic kidney disease. The pathological mechanisms underlying many of these complications remain unclear [4].
Paroxysmal nocturnal haemoglobinuria (PNH) is caused by an acquired defect of protective proteins on the red cell membrane – a deficiency of glycophosphatidylinositol. It may occur in isolation or as part of a bone marrow disorder such as aplastic anemia. Only a quarter of patients have red urine (haemoglobinuria) in the morning. It may present with acute or chronic kidney disease due to haemoglobin occluding or damaging tubules and stimulating interstitial fibrosis [5]. It may be treated with eculizumab , a monoclonal antibody that inhibits the complement activation that attacks the red cell membrane (see Fig. 14.4).
Haemolytic uraemic syndrome (HUS) is divided into two forms: typical diarrhoea-positive (D+ HUS) and atypical (aHUS).
D+ HUS is the consequence of an infection, usually due to Escherichia coli subtype O157 . The infection is typically contracted from undercooked beef, contaminated dairy products or contact with the organism in the environment such as petting farms.
The E. coli produces a shiga-like toxin, also known as verocytotoxin , which damages endothelial cells, activates neutrophils and triggers the coagulation pathway. This consumes platelets and causes a microangiopathic haemolytic anaemia (MAHA) (Fig. 13.3).
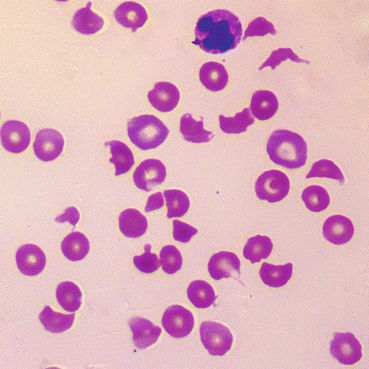
Fig. 13.3
Blood film showing microangiopathic haemolytic anaemia. There are large numbers of fragmented and abnormally shaped red cells called schistocytes indicating damage to red cell membranes, and low numbers of platelets
Microthromi occlude the glomerular capillaries, causing acute kidney injury (Fig. 13.4).
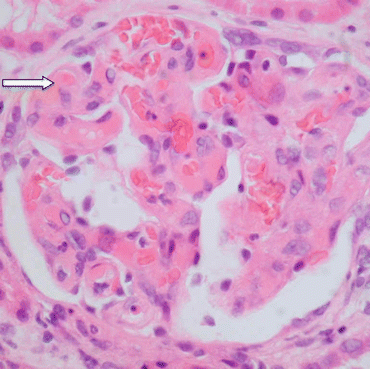
Fig. 13.4
Section of a glomerulus from a patient with HUS showing glomerular capillaries containing microthrombi and red cells (arrow). Haematoxylin and eosin ×60
Once the acute illness is passed, the glomerular microthrombi resolve and the capillaries reopen. Hence treatment of the condition is supportive, about 50 % of patients requiring dialysis, usually for up to 2 weeks (Patient 13.2).
Patient 13.2: Typical Diarrhoea-Positive (D+ HUS)
Ten-year-old Rosie was admitted to hospital with a 3-day history of diarrhea, with blood for the last 24 h. She had drunk unpasteurised milk 4 days before the diarrhoea. Serum electrolytes and creatinine were normal for her age (creatinine 50 micromol/L = 0.6 mg/dL). Haemoglobin was increased due to acute dehydration (174 g/L = 17.4 g/dL). Neutrophils were raised (17.7 × 109/L) but platelets and clotting were normal. She was given oral rehydration mixture.
Two days later serum sodium had fallen to 130 mmol/L, creatinine increased to 345 micromol/L (3.9 mg/dL). Haemoglobin had fallen to 117 g/L (11.7 g/dL) and platelets to 76 × 109/L. Stool culture grew Escherichia coli O157.
Over the next 5 days her creatinine rose to 975 micromol/L (11.0 mg/dL) and she commenced peritoneal dialysis, which continued for 13 days. Her haemoglobin fell further to 74 g/L (7.4 g/dL) and the film showed fragmented red cells typical of microangiopathic haemolytic anaemia. She was given a blood transfusion.
Two months later her creatinine had returned to her normal level and she has since remained well.
aHUS is atypical in that there is usually no or only mild diarrhoea and no E. coli infection. Hypertension is common and can be severe. Up to 70 % progress to end-stage kidney failure and there is a high risk of recurrence after a kidney transplant.
The condition can present at any age, 60 % in childhood. It may be familial and up to 80 % of cases have genetic abnormalities of complement regulation (Patient 13.3).
When the condition involves other organs, notably the nervous system with seizures and coma, it is termed thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS). Patients with TTP classically have severe deficiency of ADAMTS13 , the von Willebrand factor -cleaving protease. ADAMTS13 normally breaks down multimers of von Willebrand factor, reducing their clotting activity [6].
Treatment traditionally involved plasmapheresis and FFP infusions but had a rate of dialysis or death of 40 %. The monoclonal antibody eculizumab is now used because the condition, like PNH, is mediated by the complement cascade (see Sect. “Complement C3 and C4” in page 202).
Patient 13.3: Atypical HUS (aHUS)
Peter, aged 5, was admitted with a 3-day history of vomiting, jaundice and abdominal cramps without diarrhoea. He was pale, icteric and had petechiae on his limbs and trunk. Blood count confirmed low haemoglobin with fragmented red blood cells on the film. Serum lactate dehydrogenase and creatinine were raised (Fig. 13.5).
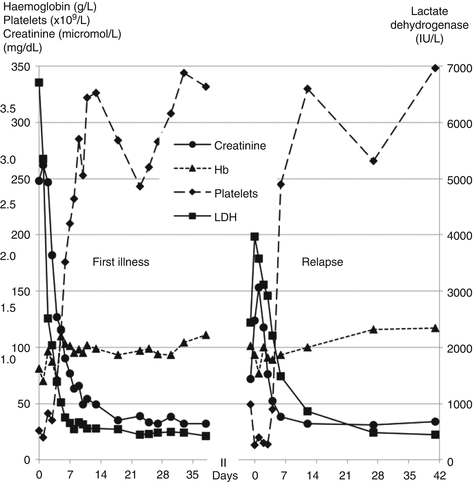
Fig. 13.5
Patterns of creatinine, blood count and lactate dehydrogenase (LDH) during two episodes of aHUS
His older brother had had an episode of low platelets and transient anaemia at the age of 5 years, diagnosed as thrombotic thrombocytopenic purpura, and had made a full recovery.
A diagnosis of atypical HUS was made and Peter was treated with daily plasma exchange. His kidney function and blood count recovered quickly. He was maintained on regular plasmapheresis and then eculizumab infusions every 2 weeks.
Genetic studies identified a mutation of the CD46 membrane cofactor protein gene in both Peter and his brother. This cofactor degrades complement components C3b and C4b, interrupting the complement cascade. Loss of its function leads to unregulated formation of the complement membrane attack complex that causes endothelial cell damage (see Fig. 14.4).
Eculizumab infusions were discontinued after 6 months but he was admitted 9 months later with a recurrence of HUS (Fig. 13.5). This responded rapidly to eculizumab alone and he remains on 3 weekly infusions.
Acid, Base, Bicarbonate and Total CO2
A simple indicator of acid-base balance in kidney disease is the concentration of bicarbonate in a venous serum sample. The laboratory may measure the serum total CO2 content instead of bicarbonate. Total CO2 includes bicarbonate ions, dissolved CO2 and carbonic acid. Bicarbonate comprises about 95 % of the total CO2 content.
Acid is mainly excreted in the urine as ammonium ions (NH4 +) . The smell of stale urine comes from the breakdown of ammonium chloride and urea to ammonia. In Roman times, stale urine was collected and used for cleaning and for whitening teeth.
As the number of working nephrons declines, ammonium excretion per remaining nephron increases. This is mediated through increased angiotensin production by the kidney. When GFR falls below about 45 ml/min/1/73 m2, total ammonium excretion starts to fall. Initially, excess H+ ions are buffered by bicarbonate in the extracellular fluid and by tissue buffers and bone. But as GFR falls further, these buffers become saturated, metabolic acidosis develops and serum bicarbonate falls.
The acidosis is not only a consequence of the loss of GFR; it also contributes to its progression [7]. The risk of GFR declining is associated with the level of serum bicarbonate, the lower the level the greater the risk. This association is found over a range of serum bicarbonate that includes normal values, that is above 22 mmol/l.
Randomised controlled trials of patients with CKD stages 3 or below and serum bicarbonate <24 mmol/L have shown that treatment with oral sodium bicarbonate or a diet high in alkali-producing fruits and vegetables significantly reduces the rate of decline in GFR and the chances of starting dialysis over the following 2–3 years.
The mechanism of this important effect is mediated by a reduction in production of angiotensin II in the kidney, measured by the level of angiotensinogen in the urine [8].
Bicarbonate treatment also increases protein intake and reduces the impact of acidosis on protein breakdown. Treated patients have better nutritional status, such as mid-arm circumference and serum albumin [9]. Sodium bicarbonate does not cause signs of sodium overload such as oedema or high blood pressure; in this respect, sodium bicarbonate behaves differently to sodium chloride.
Increasing dietary acid intake in the form of two or more cola drinks a day, which contain phosphoric acid, is associated with a doubling of the risk of chronic kidney disease [10].
Vitamin D, Minerals, Bones and Blood Vessels
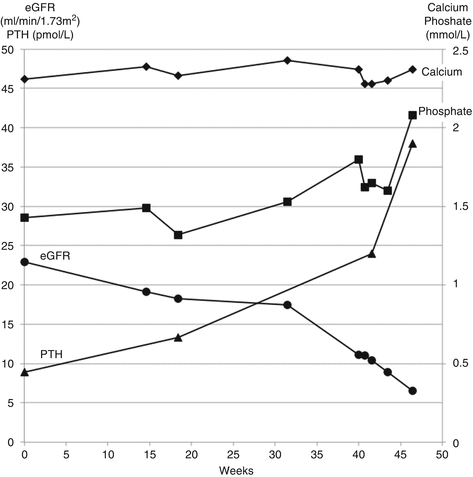
Serum phosphate is derived predominantly from the catabolism of phosphorylated proteins. As GFR declines, the excretion of phosphate decreases. Eventually excretion fails to balance production and serum phosphate increases. This is exemplified in Patient 13.4.
Patient 13.4: Development of Abnormal Mineral Metabolism in Chronic Kidney Disease
Mr. Jordan, a fit 65-year-old white man, had IgA nephropathy. His eGFR declined by more than 15 ml/min/1.73 m2/year and he showed typical changes in serum calcium, phosphate and parathyroid hormone (PTH) (Fig. 13.6). He did not take any calcium or vitamin D supplements during this time.