Fig. 4.1
An overview of the systems affecting renal blood flow and metabolism in sepsis
The renin–angiotensin–aldosterone system (RAAS) is activated during sepsis and hypotension. Renin release is stimulated by the fall in renal perfusion and is also influenced by prostaglandins, the sympathetic nervous system, and the kallikrein system. Angiotensin II may potentiate norepinephrine release, but also stimulates production of vasodilating prostaglandins, kallikrein, and nitric oxide (NO). Angiotensin II constricts efferent arterioles, thereby reducing renal blood flow but increasing filtration fraction. Besides its vascular effects, angiotensin II may also contract mesangial cells, reducing the surface area available for filtration.
The production of renal prostaglandins depends on the availability of arachidonic acid and phospholipase (PLA2). Activation of PLA2 and increased production of arachidonic acid metabolites can be triggered by several factors such as endotoxin, cytokines, platelet activating factor (PAF), tissue hypoxia, reactive oxygen species (ROS), and angiotensin II. Prostaglandins interact in a complex fashion with other regulatory systems, since they may mediate renin secretion, reduce catecholamine actions, and contribute to the release of vasopressin. Normal renal hemodynamics does not depend on the vasoactive prostaglandins. During a fall in cardiac output, however, vasodilating prostaglandin E2 and prostacyclin PGI2 are released. Conversely, activation of the lipoxygenase pathway during sepsis yields leukotrienes having vasoconstrictive properties, which adversely interfere with the filtration coefficient. Their blockade may be beneficial for renal perfusion and function. Inhibition of the cyclooxygenase-derived compounds by non-steroidal anti-inflammatory drugs (NSAIDs) reduces renal blood flow during experimental sepsis, suggesting a predominantly vasodilating effect of prostaglandins generated via cyclooxygenase in the kidney.
The balance between endothelin and NO may play a critical role in renal perfusion during sepsis. Endothelin is a potent renal vasoconstrictor and expression of endothelin receptors is increased during sepsis. The kidney produces endothelin upon stimuli including endotoxin and tumor necrosis factor (TNF)-α. Endothelin may constrict afferent more than efferent vessels, which decreases renal blood flow and glomerular filtration.
During sepsis, morphological and functional alterations of the renal microcirculation have been observed, involving vasoconstriction and capillary clotting [21, 22]. Oxidative stress and the NO-system have been described as major contributors to these phenomena. ROS can injure the endothelium, damage the glycocalyx, and impair the endothelium-dependent vasoreactivity [18, 23, 24]. NO is formed by enzymatic cleavage from L-arginine by NO-synthetase. Especially NO derived from endothelial NO-synthetase (eNOS) plays an essential role in maintaining the renal vascular homeostasis. It dilates afferent vessels in response to shear stress and angiotensin II-mediated vasoconstriction, and inhibits tubuloglomerular feedback, increasing renal blood flow. It also inhibits platelet aggregation and leukocyte activation. eNOS-derived NO thus regulates vascular tone, influences the filtration coefficient, mediates the effects of macula densa stimulation, defends medullary oxygen supply, and mediates pressure-induced sodium excretion.
On the other hand, activation of inducible NO-synthase (iNOS) has been shown to promote renal injury. Endotoxin induces the activation of iNOS, leading to the formation of intracellular ROS and reactive nitrogen species (RNS) [25, 26]. Intracellular ROS and RNS are able to damage mitochondria, leading to apoptosis. In addition, iNOS-derived NO enables shedding of tubular epithelial cells by cytokines [27]. In an attempt to mitigate renal injury caused by iNOS, septic patients were treated with an NO-synthetase inhibitor. Although this treatment increased the percentage of shock reversal, it did not change the incidence of AKI [28].
Adenosine also plays a role in the pathophysiology of sepsis-associated AKI. During stress caused by inflammation and hypoxia, adenosine triphosphate (ATP) can be released from the cell, stimulating inflammation by attracting phagocytes and activating inflammasomes [29]. The enzyme alkaline phosphatase can convert ATP to adenosine, which has anti-inflammatory and tissue-protective effects [30]. A small randomized controlled trial of alkaline phosphatase in patients with sepsis showed significant improvement of a composite endpoint consisting of creatinine clearance and requirement of renal replacement therapy [31].
4.2.3 Cell Signaling and Inflammation
During sepsis, multiple cell lines, cytokines, vasoactive substances as well as the coagulation and the complement cascade participate in the inflammatory response to infection [32]. Essential for host defense is the recognition of molecular patterns by so-called toll-like receptors (TLRs) localized on the membrane of different cell types. TLRs recognize molecules derived from bacteria, viruses, fungi, and parasites known as pathogen-associated molecular patterns (PAMPs). TLRs are also able to recognize molecules released during tissue injury, known as damage-associated molecular patterns (DAMPs). In the kidney, TLR-2 and TLR-4 are expressed both in endothelial, glomerular, and epithelial cells of Bowman’s capsule and the proximal and distal tubules [33]. Activation of TLRs triggers a series of events leading to the expression of pro-inflammatory genes [34]. Moreover, TLRs can be upregulated in the presence of interferon-γ and TNF.
Several studies in animal models of sepsis have demonstrated the role of TNF in the development of acute kidney injury: mice lacking a TNF receptor are resistant to endotoxin-induced acute kidney injury and TNF-receptor negative kidneys are resistant to endotoxin-induced damage when transplanted into TNF-positive mice [32]. In human sepsis, elevated serum levels of two soluble TNF-receptors are strongly associated with the development of acute kidney injury [35]. TNF is believed to induce apoptotic cell death through intracellular signaling. Renal tubular cells express cell surface ‘death receptors’ of the TNF superfamily, including TNF-receptor 1, Fas and FN14 receptor. TNF, TNF-like weak inducer of apoptosis (TWEAK) and Fas ligand can all activate caspases (cysteine-dependent aspartate directed proteases), enzymes with the ability to initiate apoptosis [36]. In a cascade-like manner, caspases activate other target caspases by cleaving away prodomains, ultimately activating effector caspases such as caspase-3, which cleaves multiple structural molecules, resulting in the morphologic changes that define apoptosis [37]. Treatment with a caspase-3 inhibitor was demonstrated to have a renoprotective effect in mice [38]. Caspase-1 has the ability to activate interleukin-1β and interleukin-18, cytokines playing an important role in the inflammatory process occurring during septic acute kidney injury. Blocking the activation of these cytokines also had a renoprotective effect in mice [39]. Moreover, in a murine sepsis model, combined inhibition of IL-1 and IL-18 protected animals against a lethal challenge with endotoxin [40].
Inflammation is an important component of both the initiation and extension of injury in AKI [41]. Endothelial injury stimulates the recruitment of leukocytes to the renal tissue and promotes leukocyte adhesion and migration. Leukocyte adhesion is mediated by adhesion molecules known as selectins, of which intercellular adhesion molecule-1 (ICAM-1) appears to be essential to the transendothelial migration of leukocytes [42]. The low microcirculatory blood flow observed during sepsis further facilitates the interaction between inflammatory cells and the endothelium through prolonged exposure to leukocytes and inflammatory mediators, which amplify the local inflammatory response and renal injury.
Coagulation plays an important role in the pathogenesis of sepsis. Inflammatory mediators such as endotoxin and cytokines may activate the coagulation cascade, which results in thrombin formation. Thrombin itself is a proinflammatory agent with the ability to activate inflammatory cells, which in turn produce cytokines, thus promoting an amplifying loop. The literature on the role of the coagulation cascade in the pathogenesis of acute kidney injury is scarce. Recently, the contribution of microparticles to various pathophysiologic processes has gained interest. Microparticles are small lipid bilayer vesicular bodies originating from activated or apoptotic cells. They often carry phosphatidylserine in their outer layer, which may act as a surface for activated clot formation. In addition, microparticle bound coagulation factors such as tissue factor may initiate coagulation during sepsis. A recent study among patients with severe sepsis showed that patients with renal injury had significantly increased numbers of circulating microparticles [43]. This finding suggests that activation of the coagulation pathway also plays a role in the pathogenesis of septic acute kidney injury.
4.2.4 Tissue Injury and Apoptosis
Historically, renal tubular epithelial cell necrosis was regarded as the characteristic lesion in septic AKI. However, analysis of kidney biopsies from patients who died of septic shock showed limited evidence for acute tubular necrosis, but did show tubular and glomerular cell apoptosis and infiltration of monocytes [44]. During experimental sepsis, glomeruli and interstitium are also infiltrated by activated neutrophils [32, 45]. Tubular apoptosis rather than necrosis may thus prevail in vulnerable proximal tubules. However, the relevance of apoptosis to organ dysfunction has been debated [44].
4.3 Ischemia and Reperfusion (I/R)
The I/R injury of the kidney mainly occurs in clinical conditions such as cardiovascular surgery, shock, trauma, resuscitation, and other situations associated with hypotension, low cardiac output or both [46]. The time interval of complete blood flow interruption that leads to renal injury in animals is 30–45 min, but in humans the period can be longer, as observed during vascular and renal surgery [47]. Reperfusion during shock resuscitation stimulates inflammatory factors, therefore, I/R injury shares many pathways with the pathogenesis of AKI in inflammatory states.
4.3.1 Hemodynamics
Severe renal hypoperfusion leads to ischemia, which may only be partially reversed during reperfusion [13]. However, in the absence of other risk factors, near 100 % occlusion of the renal circulation is required to cause significant ischemic renal injury [48]. Doppler and contrast-enhanced ultrasonography yield an index of renal vascular resistance and regional perfusion at the bedside and show marked increases in resistance in patients at risk for developing AKI [9, 10, 13, 14, 49].
When injured tubules reabsorb less sodium and water, this results in a negative feedback to the RAAS system at the macula densa and activation of the tubuloglomerular feedback, causing afferent vasoconstriction contributing to a sustained fall in perfusion and filtration. In particular the blood flow to the cortico-medullary junction, an area with baseline supply dependence for oxygenation, can remain compromised during reperfusion. The microvasculature in this region becomes congested due to interstitial edema, red blood cell trapping, leukocyte adherence, and extravasation upon reperfusion [50]. A fall in renal blood flow due to microvascular injury and tubuloglomerular feedback, a rise in the pre- to post-glomerular capillary resistance ratio and increased tubular luminal pressure opposing filtration across the tubular epithelium can all contribute to the low glomerular filtration following I/R [51].
4.3.2 Endothelial and Vascular Smooth Muscle Injury
Endothelial and vascular smooth muscle cells are damaged by ischemia, which contributes to vascular dysfunction [52, 53]. This includes diminished capability to autoregulate vascular tone following endothelial dysfunction. The endothelium-dependent relaxation to acetylcholine may be less effective in defending against constriction of the renal artery after I/R, and this may be associated with increased endothelin and impaired NO production by eNOS, in part downregulated by iNOS [54, 55].
4.3.3 Tubular Injury
Renal tubular cells are polarized, the apical and basolateral side having different composition and function. The tight junction separates the apical and basolateral membrane and provides a barrier to parallel transport of water and solutes. The adherens junction is located directly basal to the tight junction and contributes to cell polarity [56]. ATP depletion disrupts both the tight and adherens junction, leading to loss of cell polarity, increase in permeability and back-leak of glomerular filtrate. Prolonged ATP depletion leads to disruption of the actin cytoskeleton, collapse and detachment of tubular cells with shedding in the tubular lumen and excretion into the urine [56].
4.3.4 Cell Signaling and Inflammation
Factors which may trigger inflammatory injury after renal I/R include activation of the complement system, adherence of the membrane attack complex, microvascular thrombosis, generation of ROS, and stimulation of tubular NFκB [34]. In addition, histones derived from dying cells trigger TLRs, thereby contributing to the perpetuation of AKI [57]. These processes lead to the generation of multiple cytokines and pro-inflammatory molecules that mediate inflammatory injury after I/R [58–61]. In response to endothelial activation, inflammatory mediators, cytokines, and chemokines induce rolling, sticking, and infiltration of activated neutrophils to the interstitium, which contributes to interstitial edema and may limit the recovery of renal blood flow [50, 60]. Cytoskeletal alterations may be involved in the transmigration of neutrophils via loosened intercellular junctions. After mild I/R of the kidneys, endotoxin exposure and neutrophil priming can further contribute to AKI, and as a consequence, retention of activated neutrophils and further release of proteases and ROS. Following neutrophil infiltration, a monocyte/macrophage infiltrate may become apparent up to 24 h after renal I/R, which may be involved in tissue repair [62] or conversely in development of fibrosis and chronic renal injury [53, 63].
4.3.5 Tissue Apoptosis, Necrosis, and Repair
Apoptotic and necrotic forms of cell death coexist in I/R kidneys [36, 64, 65; Fig. 4.2]. The relative contribution of the two types of cell death depends on the severity of the injury. The degree of cellular ATP and GTP depletion may play a crucial role in determining the mode of cell death [64, 66]. Survival mechanisms may allow the cells to produce growth factors such as hepatocyte growth factor (HGF) that may regenerate proximal and distal tubules [65, 67–71]. Collectively, a range of factors may promote inflammation, apoptosis, and ultimately fibrosis after renal injury, as opposed to tubular regeneration, redifferentiation, and full restoration of renal function [71].
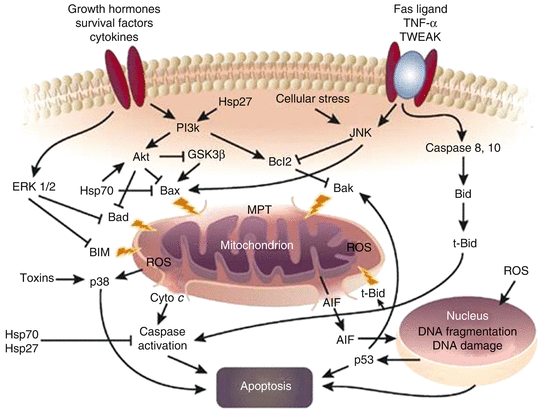
Fig. 4.2
Interaction between inflammation and apoptosis (Reproduced with permission from Nature publishing Group [36]). Abbreviations: AIF apoptosis-inducing factor, Akt protein kinase B, Bax, Bad, Bak, and truncated or t-Bid are proapoptotic BCL2 proteins, Bcl2 antiapoptotic BCL2 protein, Cyto c cytochrome c, ERK1/2 extracellular regulated kinase 1/2, GSK3b glycogen synthase kinase 3-b, Hsp heat stress protein, JNK Jun-N-terminal kinase, MPT outer mitochondrial membrane pore transition, p38 mitogen-activated protein kinase, p53 a tumor-suppressor protein, PI3k phosphatidylinositol 3-kinase, ROS, reactive oxygen species generated by mitochondria or other sources, TNF–α tumor necrosis factor-α, TWEAK TNF-related weak inducer of apoptosis. ERK1/2 and JNK are stress-regulated kinases, Hsp70 is heat stress protein 70 kDa an antiapoptotic protein, Hsp27 is heat stress protein 27 kDa is a TNF family member, BID stands for BH3 interacting-domain death agonist. It is a pro-apoptotic member of the Bcl-2 protein family
4.4 Nephrotoxicity
Nephrotoxin exposure accounts for up to 25 % of the cases of AKI in the intensive care unit [72]. Nephrotoxicity may be promoted by factors relating to the patient, the kidney and the ingested toxic substance [73]. Critically ill patients are vulnerable to nephrotoxicity due to compromised renal perfusion. The kidney itself is prone for toxicity because it takes up toxins and concentrates them in interstitium and medulla. Renal tubular cells have a high metabolic rate requiring a substantial amount of energy and their environment is relatively hypoxic, increasing the risk of hypoxic injury to the tubular cells. Toxins may potentiate renal injury induced by other causes. Moreover, hypoalbuminemia often occurs in critically ill patients, leading to a higher plasma free fraction of potentially toxic agents [74].
The most important nephrotoxins are drugs frequently used in the ICU, such as antibiotics, analgesics, and radiocontrast [72–78]. However, outpatient exposure to nephrotoxins such as heavy metals and certain herbs may also cause nephrotoxicity [79, 80] (Table 4.1). All compartments of the kidney can be injured by toxic substances: renal vessels, glomeruli, tubular system, collecting ducts, and interstitium (Table 4.2). The pathophysiologic mechanisms of the injury to the different compartments of the kidney are briefly described.
Table 4.1
Groups of nephrotoxic agents
Antimicrobial agents | Lead |
Aminoglycosides | Mercury |
Antiviral agents | Uranium |
Amphotericin B | Herbal agents |
Quinolones | Taxus |
Sulfonamides | Euphorbia |
Sulfadiazine | Immunosuppressives |
Analgesics | Calcineurin inhibitors |
NSAIDs | Tacrolimus |
Selective COX-2 inhibitors | Osmotic agents |
Chemotherapeutics | Dextrans |
Ifosfamide | Immunoglobulins |
Methotrexate | Mannitol |
Platins | Starches |
Contrast agents | Vasodilators |
Radiocontrast | Angiotensin converting enzyme inhibitors |
Gadolinium | Angiotensin receptor blockers |
Heavy metals | Vasopressors |
Bismuth | Norepinephrine |
Cadmium | Vasopressin |
Copper | Lead |
Table 4.2
Pathophysiologic mechanisms of nephrotoxicity
Impact on hemodynamics | Cisplatin |
Norepinephrine | Ifosfamide |
Vasopressine | Adefovir |
NSAIDs | Cidofovir |
Angiotensin converting enzyme inhibitors | Foscarnet |
Angiotensin receptor blockers | Cocaine |
Radiocontrast | Osmotic agents |
Amphotericin B | Mannitol |
Small vessel injury | Immunoglobulins |
Thrombotic microangiopathy | Dextrans |
Calcineurin inhibitors | Starches |
Chemotherapeutics | Urinary obstruction |
Platelet aggregation inhibitors | Crystal precipitation |
Interferon | Sulfonamides |
Quinine | Quinolones |
Cocaine | Nitrofurantoin |
Vasculitis | Acyclovir |
Allopurinol | Indinavir |
Penicillamine | Methotrexate |
Phenytoine | Triamterene |
Hyaline arteriolopathy | Myoglobin precipitation |
Calcineurin inhibitors | Antibiotics |
Cholesterol emboli | Neuroleptics |
Thrombolytic agents | Illicit drugs |
Heparin | Statins |
Warfarin | Retroperitoneal fibrosis |
Glomerular injury | Dopamine agonists |
Minimal change nephropathy | Ergot alkaloids |
Lithium | Interstitial nephritis |
NSAIDs | Antibiotics |
Interferon | Beta-lactams |
Penicillin | Quinolones |
Quinolones | Macrolides |
Focal and segmental glomerulosclerosis | Rifampin |
Pamidronate | Sulfonamides |
Lithium | Tetracyclines |
Heroine | Antivirals |
Interferon | Acyclovir |
Membranous glomerulonephritis | Indinavir |
NSAIDs | Cocaine |
Penicillamine | NSAIDs |
Captopril | Diuretics |
Tubular injury | Thiazide diuretics < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|


