Fig. 16.1
Inherited endocrine condition related to mineralocorticoid excess. The picture shows the molecular pathways involved in dysregulation of NaCl homeostasis located in the distalrenal tubules. AME apparent mineralocorticoid excess; GRA glucocorticoid-remediable aldosteronism; PHA2 pseudohypoaldosteronism type 2; MR mineralocorticoid receptor; WNK with-no-lysine (K) kinase 1,4; ROMK renal outer medullary potassium channel; ENaC epithelial sodium channel; KLHL3 kelch-like 3; CUL3 cullin 3; Sgk1 serum glucocorticoid kinase 1; Nedd4–2 ubiquitin-protein ligase; deubiquitylating enzyme Usp2–45; 14–3-3 proteins; NHERF2 Na+/H+ exchange regulating factor 2; PDK1, 2 phosphoinositide-dependent kinases 1 and 2; + activation; − inhibition. (Reprinted from Melcescu and Koch [1])
When considering the current obesity epidemic, with its many hormonal derangements (e.g., deficiencies of testosterone, vitamin D, growth hormone), the hitherto estimated prevalence rates of approximately 10 % for endocrine HTN are likely to be an underestimate. Potential nonadrenal causes of endocrine HTN include excess production of growth hormone (acromegaly), thyroid hormone, and parathyroid hormone, as well as insulin resistance, hypothyroidism, and overstimulation of central MRs [1, 5–40]. These “nontraditional” forms of endocrine HTN will not be discussed in detail in this chapter.
Cutoffs of systolic and diastolic blood pressure (BP) for defining HTN along with reference ranges for hormonal assays among various patient populations are all important to consider when applying existing or emerging data to clinical patient scenarios. HTN guidelines by various societies have been recently revisited [41–46]. The American Society of Hypertension/International Society of Hypertension (ASH/ISH) guidelines reaffirm the traditional threshold of 140/90 mmHg as a cutoff for defining an elevated BP and state that individuals with a BP of 140–159/90–99 mmHg and no other risk factors are considered at low risk. On the other hand, the Joint National Committee (JNC 8) suggests a systolic blood pressure (SBP) of 150 mmHg as a worrisome threshold, especially for those 80 years and older. For the diastolic BP goal, both the ASH/ISH and JNC 8 guidelines view 90 mmHg as the cutoff; the exception are diabetic hypertensives for whom the cutoff is 85 mmHg. From epidemiologic studies and drug trials, there appears no sound evidence to continue recommending a BP of 140/90 mmHg as the cutoff to initiate antihypertensive treatment; moreover, any increased benefit from additional antihypertensive medications can be nonexistent or negligible, particularly in the face of increased likelihood of adverse effects.
Nevertheless, both the JNC 8 panel and ASH/ISH group recommended a treatment threshold of 140/90 mmHg for adults with diabetes or chronic kidney disease (CKD), based on expert opinion due to lack of randomized controlled evidence. For initial HTN therapy, a thiazide-type diuretic or calcium channel blocker was recommended, and in nonblack patients, the use of an angiotensin-converting-enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) . The latter recommendation is based on the large-scale ALLHAT (antihypertensive and lipid-lowering treatment to prevent heart attack trial). In the era of patient-centered medicine, it is important to remain flexible with these guidelines and use them on an individualized basis. Hopefully, future data (2018 and later) from more randomized trials such as the systolic blood pressure intervention trial (SPRINT) will clarify whether a BP of less than 140 mmHg or less than 120 mmHg is more beneficial for nondiabetic adults of age 50 years and older.
Case Finding and Screening
Resistant HTN represents an important case finding feature for endocrine HTN that is often associated with obesity, sleep apnea, and reduced renal function among other conditions. With the current obesity epidemic, approaching an endocrine assessment of HTN can be difficult [47]. There are many (neuro)endocrine alterations in obese patients with and without sleep apnea, including an increase in leptin, cortisol, insulin, and aldosterone levels and a decrease in growth hormone, prolactin, and testosterone [48]. Among National Health and Nutrition Examination Survey (NHANES) participants with CKD, only 37 % had a BP < 130/80 mmHg, leaving 63 % with a BP higher than 130/80 mmHg. In ALLHAT, 34 % of patients taking at least two antihypertensive drugs continued to have uncontrolled HTN after 5 years of follow-up [49]. For patients with truly resistant HTN, the new recommendations include withdrawing medications that do not lower BP, and consideration of a mineralocorticoid antagonist, amiloride, or doxazosin as alternatives. Furthermore, renal denervation or baroreceptor stimulation should be considered if optimal drug therapy is deemed ineffective. These invasive approaches should be reserved for patients with clinic values > 160 mmHg SBP or > 110 mmHg and with BP elevation confirmed by ambulatory BP monitoring. In treating resistant hypertensive patients, renal nerve ablation is associated with a decrease in plasma noradrenaline but not in renin [50].
Of note, the exact prevalence of resistant HTN in patients with CKD remains unknown, but may reach to 30 % of all patients with uncontrolled HTN in the USA [51, 52]. Large-scale population-based studies such as the US NHANES suggest that 12 % cases of HTN are resistant to pharmacological therapy [53]. Importantly, apparent treatment resistance is often related to poor adherence to antihypertensive therapy, which is reflected by findings that about 40 % of patients with newly diagnosed HTN discontinue their treatment during the first year [52]. In all patients with an albumin excretion rate of > 30 mg/24 h, the use of an ACE inhibitor or ARB is recommended [54]. A recent review of the evidence on the role of dietary salt and potassium intake in cardiovascular health and disease revealed that modest salt restriction while increasing potassium intake serves as a strategy to prevent or control HTN and decrease cardiovascular morbidity and mortality [55].
CKD affects approximately 15 % of the adult population, and intensive BP lowering apparently can protect against kidney failure events in patients with CKD, especially among those with proteinuria [56]. A systematic review and meta-analysis suggest that a 10 mmHg reduction in SBP might result in an overall reduction of 22 % in the risk of kidney failure [56].
One important question in this regard is when to screen for secondary causes of HTN, including endocrine HTN . HTN in young patients and refractory HTN (characterized by poorly controlled BP on > 3 antihypertensive drugs) should alert the physician to screen for secondary causes. The clinician should carefully screen for cardinal signs and symptoms of CS, hyper- or hypothyroidism, acromegaly, or pheochromocytoma . The importance of endocrine-mediated HTN resides in the fact that, in most cases, the cause is clear and can be traced to the actions of a hormone, often produced in excess by a tumor such as an aldosteronoma in a patient with HTN due to PA . More importantly, once the diagnosis is made, a disease-specific targeted antihypertensive therapy can be implemented, and in some cases, surgical intervention may result in complete cure, obviating the need for lifelong antihypertensive treatment.
Clinical Diagnosis of Endocrine Hypertension
A detailed medical history and review of systems should be obtained . The onset of HTN and the response to previous antihypertensive treatment should be determined. Consideration should be given to compliance to prescribed antihypertensive regimen. A history of target organ damage (i.e., retinopathy, nephropathy, claudication, heart disease, abdominal or carotid artery disease) and the overall cardiovascular risk status should also be explored in detail .
As in other causes of HTN, the clinician should question the patient about dietary habits (salt intake, etc.); weight fluctuations (recording exact dry weights, etc., in patients with chronic renal disease); and use of over-the-counter drugs and health supplements, including teas and herbal preparations, recreational drugs, and oral contraceptives. Moreover, a detailed family history may provide valuable insights into familial forms of endocrine HTN. The review of systems should include disease-specific questions. Most patients with HTN due to pheochromocytoma are also symptomatic. Symptoms may include headaches, palpitations, anxiety-like attacks, and profuse sweating, similar to symptoms of hyperthyroidism. Pheochromocytoma, CS, and other endocrine conditions may represent secondary causes of diabetes mellitus [57] .
Patients with CS often complain of weight gain, insomnia, depression, easy bruising, and fatigue. Acne and hirsutism (in women) can also be observed. The challenge these days is to recognize patients with evolving CS among the many obese and often poorly controlled diabetic individuals. Primary hyperaldosteronism is manifested by mild to severe HTN [58–60]. Hypokalemia can be present but it is not a universal finding, and there is normokalemic PA . Polyuria, myopathy, myoglobinuric acute renal failure, and cardiac dysrhythmias may occur in cases of severe hypokalemia [61]. Hypokalemia is associated with kidney function impairment in patients with PA [62, 63], because chronic hypokalemia could induce chronic renal failure through characteristic tubulointerstitial damage consisting of vacuolization of epithelial tubular cells and interstitial fibrosis [64]. In the study of Takanobu and colleagues [65], however, preoperative serum potassium levels did not differ significantly among untreated PA patients. This result suggests that excretion of potassium could have already declined in patients with progressive renal damage, such as epidermal growth factor receptor (eGFR) < 60 mL/min/1.73 m2 .
Many adrenal tumors are today being discovered incidentally during imaging procedures. The challenge for health-care providers is to identify those lesions that may be potentially harmful by either excessive hormone secretion, compressive effects on other structures adjacent to the adrenal glands, or spreading metastases . When caring for a patient with impaired renal function and an incidentally discovered adrenal tumor, screening for excessive hormone secretion from the tumor may be challenging, as many of our contemporary hormonal cutoff values, including stimulation and suppression tests, are based on patient populations with normal renal function. (Fig. 16.2) In both patient groups, those with and those without normal renal function, the transition from normal to subclinical and to finally full-blown clinical hormonal oversecretion often is difficult to recognize. When evaluating a patient with impaired renal function and an adrenal incidentaloma, one should attend to unexplained hypokalemia, typical cushingoid features, signs of hyperandrogenism, spells including headaches, palpitations, sweats, severe or refractory HTN, and familial syndromes that are associated with adrenal tumors (e.g., von Hippel–Lindau (VHL) syndrome, multiple endocrine neoplasias) .
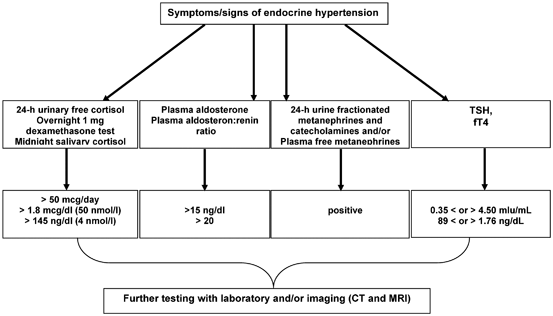
Fig. 16.2
Testing algorithm for endocrine hypertension. (Reprinted with permission from the App “Endocrine hypertension,” June 2014, by Melcescu and Koch, of www.endotext.org, the FREE online comprehensive, authoritative, constantly updated web-based Endocrinology Textbook)
In patients with a positive screening test, subsequent confirmation by various testing modalities is necessary. The most used investigations are listed in Table 16.1 .
Table 16.1
Tests for diagnosing the most prevalent forms of endocrine hypertension
Cushing’s syndrome |
|---|
1 mg dexamethasone suppression test Midnight salivary cortisol, diurnal rhythm ACTH-dependent (5–10 %; ACTH 20 ng/l) High-dose DST or CRH test If positive, then pituitary MRI and/or BIPSS If negative, then chest/abd MRI and/or somatostatin scan ACTH-independent (90–95 %; ACTH 10 ng/l) Adrenal CT or MRI |
Hyperaldosteronism |
Plasma aldosterone and direct renin Salt suppression test Positive if aldosterone excretion 12–14 µg/day while urine Na 200 mEq/day Adrenal CT or MRI Adrenal vein sampling |
Pheochromocytoma |
Plasma-free and/or urine-fractionated metanephrines Anatomic imaging (CT/MRI): a. abd/pelvis if negative then b. chest/head and neck Functional imaging: [123/131]Iodine-MIBG Specific PET ([18F]FDA, [18F]FDOPA) Nonspecific PET ([18F]FDG) Genetic testing |
Primary Aldosteronism
PA has first been described in a Polish patient with HTN and hypokalemia who experienced normotension and normokalemia after removal of an adrenocortical adenoma [66]. In patients with HTN and adrenal incidentaloma, the median prevalence of PA is estimated to be 2 %. In patients with resistant HTN, the estimated prevalence of PA is between 17 and 23 % [67]. After applying the aldosterone-to-renin ratio (ARR) not only to hypokalemic but also normokalemic hypertensives, the diagnosis of PA increased up to 15-fold [68].
The ARR, commonly used to screen for PA, has also been viewed as an index for salt sensitivity and recently has been linked to the development of CKD in a longitudinal, observational study of 698 Japanese individuals. The mean follow-up in that study was 9 years, and those people with a higher ARR more frequently developed CKD [69]. In the German Diabetes and Dialysis Study, including 1255 diabetic hemodialysis patients, the combined presence of high plasma aldosterone > 200 pg/ml and high serum cortisol > 21 mcg/dl increased the risk of sudden cardiac death as well as all-cause mortality compared to patients with an aldosterone level less than 15 pg/ml and a cortisol value less than 16.8 mcg/dl [70].
Although PA has long been viewed as a relatively benign form of HTN [71, 72], recent studies suggest that long-term exposure to high aldosterone levels might lead to cardiovascular and renal structural damage that seems to occur independent of the BP level [73, 74]. Furthermore, significant histological damage to the kidney was noted in PA patients [63, 75].
Animal Studies
Animal studies support the role of aldosterone in the progression of renal vascular disease [76–79]. In desoxycorticosterone acetate (DOCA)-salt rats, micropuncture studies have demonstrated glomerular hyperfiltration, reflecting increased glomerular blood flow consequent upon vasodilation of both afferent and efferent arterioles, and ascribed to volume expansion [80]. This renal vasodilation may be in large part via a direct mineralocorticoid effect on the vessel wall [81] and is in sharp contrast to the vasoconstrictor response to increased renal perfusion pressure in essential HTN. Studies in dogs indicate that an increase in renal artery pressure, which raises glomerular filtration rate and fractional sodium excretion, is essential in allowing the kidneys to escape from the chronic sodium-retaining action of aldosterone and to achieve sodium balance and a stable level of arterial pressure without severe volume expansion and ascites [82]. Preclinical studies in animal models clearly show that inappropriate aldosterone levels for sodium status can produce extensive renal damage [83].
Clinical Trials
Although preclinical studies indicate that aldosterone per se might cause important renal damage [76, 78], the clinical evidence supporting a direct role of this hormone as a potential contributor to renal dysfunction is limited.
Cross-sectional studies on renal function in PA have shown a high degree of variability in the prevalence of clinically relevant renal damage [84–86]. In fact, the majority of initial reports indicated that PA is less likely to cause overt renal damage [71, 72, 87, 88].
Two recent studies, a short-term [89] and a long-term follow-up [90], demonstrated that the renal dysfunction of PA is closely related with the hemodynamic adaptation of the kidney to the effect of aldosterone excess. In particular, the long-term follow-up study has shown that the condition is characterized by partially reversible renal dysfunction, in that glomerular hyperfiltration is corrected and urinary albumin excretion significantly reduced after either surgical or medical treatment of PA [89]. In agreement with the findings of studies that were conducted in more experimental settings [80–82], longitudinal studies consistently demonstrate that the hallmark of renal dysfunction in PA is reversible glomerular hyperfiltration that contributes to increase urinary albumin losses.
In a short-term study of adrenal adenoma patients after adrenalectomy, the evidence favors a major hemodynamic driver for the increased albuminuria of PA [89]. Patients with proteinuria have lower preoperative serum potassium levels than those without proteinuria [91].
This contrasts with the partial response in terms of BP and albuminuria seen in patients with higher pretreatment plasma renin levels, a possible marker for more severe renal structural damage [89].
Patients with an aldosteronoma are heterogeneous concerning mechanisms for impaired eGFR: those whose eGFR improved after adrenalectomy had lower preoperative plasma renin than those with decreased eGFR after the operation. In those patients with decreased eGFR after removal of the aldosteronoma, preoperative plasma renin was higher and not completely suppressed by the elevated aldosterone . This suggests the coexistence of a high aldosterone state due to the adenoma and an additional nonsuppressible renin state from other causes such as essential HTN. This nonsuppressible renin state would result in additional kidney damage that cannot be reversed by adrenalectomy [91, 92].
Correct interpretation of renal function in patients can be difficult using conventional eGFR before surgery, because in patients at an early stage, subtle kidney impairment might be masked by the glomerular hyperfiltration peculiar to PA preoperatively [93]. Recent studies have shown that many PA patients show a significant decline in eGFR within 6 months of follow-up without any further decrease or increase later on, because the glomerular hyperfiltration state has disappeared after treatment [62].
The occurrence of hypoaldosteronism after unilateral adrenalectomy can be a potential risk factor for postoperative development of CKD, because low aldosterone with improving HTN might decrease growth factor receptor (GFR) and lead to renal impairment [94].
Cardiac and Renal Outcomes
Excessive production of aldosterone in PA patients has been noted to lead to a higher frequency of cardiovascular events, as compared with patients suffering from essential HTN [95]. Furthermore, CKD itself has been suggested to show a significant association with risks of death, cardiovascular events, and hospitalization [96] .
Patients with aldosteronoma and left ventricular hypertrophy had lower eGFR compared to those without left ventricular hypertrophy [91]. The dysfunction of both heart and kidneys may be more closely related to other mechanisms, such as generalized endothelial dysfunction and increased oxidative stress [97, 98]. Renal damage might be underestimated in PA patients preoperatively. In the study of Takanobu, all patients with manifested CKD showed a preoperative eGFR of 60–89 mL/min/1.73 m2. Patients with preoperative eGFR of 90 mL/min/1.73 m2 did not progress to an eGFR of 60 mL/min/1.73 m2 because of higher baseline eGFR.
Utsumi et al. [99] found that the previous presence of dyslipidemia is an independent predictor for the postoperative development of CKD. Additional factors, such as older age, lower diastolic BP, and lower estimated GFR, also influence the development of CKD. In particular, aging is closely associated with declining renal function [100, 101]. Furthermore, older age might affect lower diastolic BP in patients with manifested CKD, as a result of the fall in diastolic BP after approximately 50 years of age [102] .
Diagnosis
The ARR is useful to screen for PA in patients in whom there is an expected high prevalence of PA. Such patient groups are those with resistant HTN, HTN and adrenal incidentaloma, HTN grade 2 or 3, HTN and spontaneous or diuretic-induced hypokalemia, HTN and a family history or early-onset HTN or cerebrovascular accident younger than age 40, and all hypertensive first-degree relatives of patients with PA [67]. As there is no general consensus on the ARR cutoff, sensitivity and specificity vary widely. The ARR has good within-patient reproducibility and an accuracy of 80 % for identifying patients with an aldosterone-secreting adenoma [68].
Antihypertensive drugs are the most confounding factor affecting the measurement of aldosterone and renin. Especially, MR antagonists, such as spironolactone, eplerenone, and canrenone, should be discontinued 4–6 weeks prior to screening for PA and prior to adrenal vein sampling (AVS), because these agents can lead to an increase in renin secretion and subsequently aldosterone secretion from the unaffected side (if only one adrenal gland was oversecreting aldosterone). Only half of the patients (PA or essential HTN) can be studied drug-free or on medication with minimal influence on the ARR, with approximately 20 % of patients switching antihypertensive drugs experiencing an increase in BP and several patients suffering serious adverse events requiring hospitalization [103].
In 27 uremic patients on a chronic dialysis program, there was no correlation between BP and renin. After dialysis, renin activity rose significantly [104]. In such patients on chronic hemodialysis, the renin–angiotensin system apparently still functions regarding circulatory homeostasis with challenges by volume loss or loading, as demonstrated with 1.5–2 l of intravenous saline infusion that resulted in an increase in plasma volume by 0.4 l and BP by 10–15 mmHg, but in a decrease in plasma renin activity (PRA) by 40 % [105]. In hyperkalemic patients with chronic renal failure and mild azotemia, PRA and aldosterone levels usually are lower compared to patients with normokalemia [106]. In some patients with PA and end-stage renal disease (ESRD), aldosterone excess exists for many years, if hypokalemia is “masked” by normokalemia in the setting of chronic renal failure which may become unmasked after hemodialysis when hypokalemia due to PA can develop [107]. When interpreting the ARR, one should consider the specific patient population, as elderly and/or black patients often have low PRA values which can result in high ARR while plasma aldosterone levels are rather low. Also, black individuals are more sensitive to aldosterone regarding BP elevation than white people [108]. Most individuals with a plasma aldosterone concentration less than 9 ng/dl have normal suppression after administration of fludrocortisone [109]. When assessing the ARR, direct active renin is rarely measured and standardized/compared to the traditional plasma renin activity [110, 111]. Before measuring the ARR, certain factors should be considered, as mentioned in the Endocrine Society Practice guidelines [67]. Such factors include to correct hypokalemia, to collect blood midmorning, avoiding stasis and hemolysis, maintaining the sample at room temperature and not on ice, separating plasma from cells within 30 min of collection, considering age, the levels of potassium and creatinine, the intake of estrogen-containing medications, as estrogens can increase angiotensinogen synthesis by the liver, and considering other medications that can affect the ARR. Among drugs that have minimal effects on plasma aldosterone levels are the ones listed in Fig. 16.3.

Fig. 16.3
Medications that have minimal effects on plasma aldosterone levels and can be used to control hypertension during case finding and confirmatory testing for PA. (Reprinted from Funder et al. [67], with permission from The Endocrine Society)
It is recommended to withdraw these medications at least 4 weeks before screening: spironolactone, eplerenone, amiloride, triamterene, potassium-wasting diuretics, licorice-containing products. This clearly may represent a challenge in patients with chronic renal failure and HTN.
After screening for PA with the ARR, patients should typically undergo a confirmatory test for which various procedures exist, including oral sodium loading, saline infusion, fludrocortisone suppression, and a captopril challenge. Depending on the degree of chronic renal failure and HTN, such tests may not be safe to be performed in an individual patient. This becomes especially an issue when considering the lack of data in such patient groups. In patients without PA and normal or only slightly impaired renal function, plasma aldosterone usually suppresses to less than 6 ng/dl after 2 l of intravenous saline infusion. Assessing urinary aldosterone levels is also an option in patients with normal renal function who are suspected to have PA [67]. After oral sodium loading, the urinary aldosterone should be less than 10 mcg/day.
Imaging can be performed by computed tomography (CT) or magnetic resonance imaging (MRI). A typical aldosteronoma is shown in Fig. 16.4.
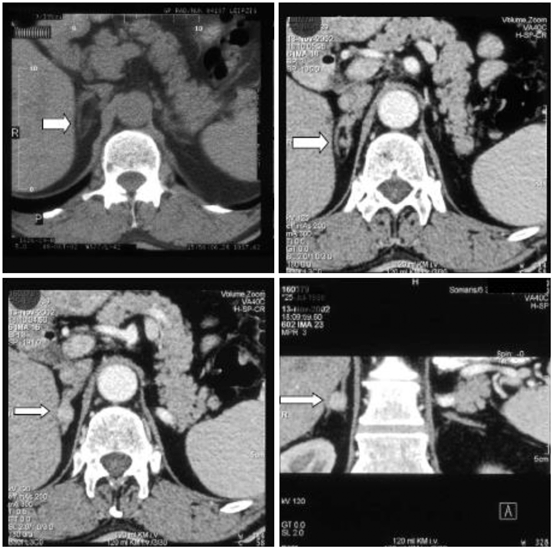
Fig. 16.4
Computed tomography scan of the adrenal glands. Left upper: without contrast, all other images with contrast, right lower image: coronary section. The white arrow points to the right adrenal adenoma in between the adrenal limbs. (Reprinted with permission from Chap. 26 “Overview of Endocrine hypertension,” December 2009, by Koch CA, Wofford MA, Ayala AR, Pacak K, of www.endotext.org, the FREE online comprehensive, authoritative, constantly updated web-based Endocrinology Textbook)
AVS should be performed in patients with PA who are considered for adrenalectomy, unless they are younger than 40 years with marked PA and have a clear unilateral adrenal adenoma, have an unacceptable high risk of adrenal surgery, are suspected to have adrenal cancer, or are proven to have familial hyperaldosteronism type 1 (glucocorticoid (GC) remediable) or type 3 (often due to germline mutations in the KCNJ5 potassium channel gene and treated with bilateral adrenalectomy). MR antagonists or amiloride should be discontinued 4–6 weeks before AVS, and AVS should best be performed in the morning, if cosyntropin stimulation is not used [112]. The selectivity index cutoff value (difference between affected and unaffected sides) should be greater than 2.0 under unstimulated conditions and greater than 3.0 during cosyntropin stimulation. In patients with PA and CKD stage III, stage IV, and stage V, AVS could be performed safely with acute kidney injury occurring in two patients [113] (see Fig. 16.5).
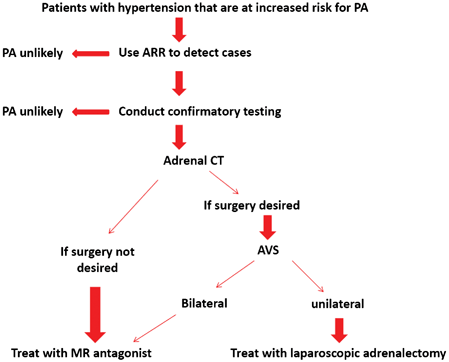
Fig. 16.5
Algorithm for the detection, confirmation, subtype testing, and treatment of PA. (Reprinted from Funder et al. [67], with permission from The Endocrine Society)
Genetic Aspects of Primary Aldosteronism
Deciphering the human genome has helped identify several disease-causing genes, including some being involved in PA, i.e., in the K-channel gene KCNJ5, encoding Kir3.4, a member of the inwardly rectifying K+ channel family [114] . The presence of these mutations in the KCNJ5 gene alter the K+ conductance/selectivity of this channel and consequently increase the Na+ conductivity (influx) with a further impact on voltage-gated calcium channels, leading to cellular proliferation in the adrenal cortex [115]. The recurrent somatic mutations (G151R, L168R) in the adrenal potassium channel KCNJ5 have been related to benign aldosterone-producing adenomas (APAs) with initial estimates, reporting almost half of APAs being associated with these mutations [115–118]. Subjects with the KCNJ5 G151E mutation have no features of APAs and hyperplasia, a different clinical course (not progressive), and excellent control of BP with spironolactone .
We now know of familial aldosteronism type 1, type 2, and type 3, although the precise molecular pathogenesis of aldosteronomas still needs to be elucidated [119–122]. FH−1 is caused by adrenocorticotropic hormone (ACTH)-dependent aldosterone secretion, GC remediable, and therefore treated with dexamethasone and/or MR antagonists, whereas FH−2 is non-GC remediable and indistinguishable from sporadic PA. FH−3 is resistant to pharmacotherapy and therefore treated with bilateral adrenalectomy. Given the heterogeneity of tumors not only between but also within one individual, it may be more prudent to analyze family members with a known gene defect and identical tumors in a whole genome- and exome-wide sequencing project for targeted genes known to be involved in cell growth regulation. Some of these patients with familial aldosteronism (overall less than 5 % of patients with PA) may require bilateral adrenalectomy to control their HTN which can occur at young age (sometimes in childhood). Exome sequencing of aldosterone-secreting adenomas has revealed somatic hotspot mutations in the ATP1A1 (encoding an Na(+)/K(+)ATPase alpha subunit) and ATP2B3 (encoding a Ca(2+)ATPase) genes [123, 124] in a small subset of tumors. Similarly, somatic mutations in CACNA1D, encoding a voltage-gated calcium channel, potentially causing increased calcium influx with subsequent aldosterone production and cell proliferation in adrenal glomerulosa cells, have been identified in less than 10 % of aldosteronomas [125] .
Therapy
Prevention of Kidney Damage by Treating Primary Aldosteronism
Adrenalectomy
Unilateral adrenalectomy can lead to a decrease in GFR as a release from the hyperfiltration state, which has to be considered a specific functional response to treatment in PA [62]. Adrenalectomy itself does not worsen kidney function, but shows the masked renal damage in PA patients postoperatively.
In animal models, MR blockade by eplerenone prevents the development of the glomerular, interstitial, and renal vascular damage caused by inappropriate mineralocorticoid-salt excess status [129, 130]. Clinically, Epstein and colleagues [131] showed MR blockade to be superior to ACE inhibition in lowering levels of proteinuria, with the combination better than either alone. In ESRD patients on hemodialysis, small pilot studies have shown that spironolactone therapy did not result in higher hyperkalemia rates [132].
The Endocrine Society Practice guidelines recommend medical management with a MR antagonist for patients who do not undergo adrenalectomy [67]. Spironolactone is a nonselective MR antagonist with an incidence of gynecomastia of approximately 7 % for doses at 50 mg/day and of more than 50 % for doses higher than 150 mg/day. Matsuda and colleagues [133] found that fecal potassium excretion in an anuric patient with PA and hypokalemia decreased while serum potassium increased after administration of spironolactone 50 mg/day. Eplerenone is a selective MR antagonist with 60 % of the potency of spironolactone and a shorter half-life but similar antihypertensive efficacy [134]. Amiloride has also antihypertensive effects in patients with PA and can be used in combination with other antihypertensives to achieve BP control . Patients with unilateral aldosteronomas should be offered unilateral laparoscopic adrenalectomy, as their HTN is cured in around 50 % of cases with many others experiencing reduction of BP values and subsequently the number of antihypertensive medications and costs [68].
Cushing’s Syndrome
Introduction
Most cases of CS are caused by exogenous GCs. Endogenous overproduction of GCs by the adrenal cortex can be the result of an ACTH-secreting pituitary tumor (approximately 80 % of cases of endogenous CS), ACTH- or corticotropin-releasing hormone (CRH)-secreting nonpituitary neuroendocrine tumor (approximately 10 % of cases of endogeneous CS), or ACTH-independent adrenal tumor [135].
HTN is a common complication of CS. HTN has a prevalence of approximately 55–80 % in adult Cushing’s patients and 50 % in children and adolescents [136–138]. GCs have a myriad of actions on multiple organ systems. GC-induced HTN results from many interacting pathophysiological pathways, which ultimately lead to an increased cardiac output (CO), total peripheral resistance (TPR) and renal vascular resistance (RVR) [136]. GCs may also influence vascular tone, BP, and electrolyte homeostasis through their mineralocorticoid activity. Indeed, free cortisol binds not only to glucocorticoid receptors (GRs) but also to MRs, and its activity at the tissue level is mediated by the microsomal enzyme 11-β-hydroxysteroid dehydrogenase (11β-HSD).
The renin–angiotensin–aldosterone system is also stimulated by GCs through increased hepatic synthesis of angiotensinogen and increased angiotensin II receptor type 1 concentrations in peripheral tissues [139, 140]. Nonetheless, patients with endogenous CS have normal or suppressed PRA [141, 142].
In patients on hemodialysis, the cortisol-to-cortisone ratio is increased due to reduced activity of 11-β-hydroxysteroid dehydrogenase [143]. Renal 11β-HSD2 expression is reduced in patients with impaired renal function, potentially causing the MR be occupied by GCs such as cortisol which may contribute to increased sodium retention in such patients [144]. Therefore, some investigators suggest making the analysis of urinary 5α-tetrahydrocortisol, 5β-tetrahydrocortisol, and tetrahydrocortisone part of the routine workup of patients with CKD and HTN [145]. Hypokalemia in obese anuric patients on chronic hemodialysis can point to CS from an adrenal tumor [146]. In this context, one should also consider the rare occurrence of apparent mineralocorticoid excess syndrome in which affected patients homozygous for missense mutations in the HSD11B2 gene, encoding 11β-HSD, have an increased ratio of urine-free cortisol to cortisone, often requiring treatment with potassium supplements and spironolactone [147].
GCs are associated with occlusive vascular disease in humans, which occurs through a myriad of effects on vascular smooth muscle, endothelial cells, myocardium and macrophages , as well as their link with obesity, HTN, dyslipidemia, and insulin resistance.
Changes in Glomerular Function
In patients with adrenal insufficiency, sodium and mineralocorticoid replacement and volume expansion were insufficient to correct the fall in renal blood flow and GFR [154, 155]. This result reflects the essential role of cortisol in the maintenance of normal renal blood flow and GFR [156].
As most GCs exhibit some mineralocorticoid effects, it has been suggested that plasma volume expansion due to sodium retention might lead to an increased GFR. However, GFR remained elevated in studies using GCs almost completely devoid of mineralocorticoid activity as well as in animal experiments with low or zero sodium intake. Thus, the GC-induced rise in GFR was not solely volume dependent [158, 160, 161].
Studies examining GC effects on glomerular vasculature are conflicting. In dogs, rats, and sheep, GCs increase renal blood flow, but this outcome is not always the case in humans [159, 161–163].
Although acute effects of synthetic or endogenous GCs increase the GFR in laboratory animals, dogs, and humans, long-term effects of CS in humans may decrease GFR as shown in the study by Haentjens and coworkers [164]. Most of the patients included in the latter study had been cured of CS, and had a decreased GFR compared to matched controls. Interestingly, the strongest predictor of GFR was duration of active disease and 4 of 18 Cushing’s patients were identified with CKD. Permanent alterations of vessel remodeling due to the chronic endogenous state of excess cortisol may have contributed to a lower GFR in these patients [164]. Two other studies demonstrated a lower creatinine clearance in Cushing’s patients with active and cured disease compared with controls [165, 166].
Serum creatinine is often used as an indirect measurement of GFR. Because muscle mass is the main source of serum creatinine and urinary creatinine elimination is constant over time, diseases affecting muscle mass influence the serum creatinine level [167, 168]. Both muscular atrophy and truncal obesity are features of CS in humans and dogs, and they have been shown to result in a decreased creatinine production rate that may be compensated by muscular hypertrophy associated with obesity [169]. In people with CS, both increased and reference range serum creatinine levels have been reported [164, 166, 169]. Patients receiving prednisone showed a rise in plasma creatinine and urinary creatinine excretion, which was probably due a catabolic effect [170]. Serum creatinine in dogs with CS was usually within or lower than the reference range [171, 172].
Effects of Glucocorticoids on Fetal Renal Development
GCs have a direct effect on growth and differentiation of a wide range of fetal tissues, especially close to term [173]. In addition, GCs can program tissues in utero and mediate effects of environmental or nutritional challenges during pregnancy , which leads to long-term consequences in later life [174, 175]. Administration of GCs affects renal function. In the fetal kidney, GCs stimulate angiotensin II, vasopressin, ACTH, and leptin receptor function, as well as metabolic enzymes, epithelial Na+ channels, Na+/K+ ATPase and Na+/H+ ion transporters and aquaporins [176, 177]. Several studies have shown that the main effect of prenatal GCs was a reduced number of nephrons, which was commonly associated with the development of HTN in later life [178, 179].
Effects of Glucocorticoids on Renal Function
Proteinuria
Urinary albumin is increased in humans, dogs, and rats treated with GCs as well as canine Cushing’s patients [180–183]. Reports on proteinuria in people with naturally occurring CS are very rare, but one study described increased urinary albumin excretion in more than 80 % of patients before treatment, which was almost completely resolved after successful therapy [184]. This finding was corroborated in a second report, which mainly involved cured subjects without an increase in microalbuminuria [164]. In general, renal proteinuria may be caused by increased glomerular filtration resulting from higher intraglomerular pressure, damage to the glomerular barrier, or decreased tubular reabsorption [185].
Chronic Kidney Disease
Excess GCs have many detrimental effects on kidney function, but whether people or dogs with CS are at risk for developing CKD is an essential issue that warrants further investigation.
In humans, the combination of Cushing’s disease and renal failure is considered to be very rare [186, 187]. However, one case-control study reported a decreased GFR in a group Cushing’s patients mainly with cured disease; 4 out of 18 patients had CKD, which was defined as a GFR < 60 mL/min/1.73 m2. The authors concluded that follow-up of renal function after treatment for CS was important because a decreased GFR may have implications for medication dosages [164].
Creatinine clearance was also lower in patients with active and cured CS compared with controls in two other studies, although these differences were not statistically significant [165, 166]. In addition, a highly significant correlation between endogenous GC production and the progression rate of chronic renal failure has been described [188].
Nephrolithiasis
Compared to the general population of industrialized countries, nephrolithiasis is remarkably common in human Cushing’s patients. Indeed, studies have shown that there is a prevalence of approximately 50 % in patients with active disease and 17–27.3 % in cured patients [164, 166]. Importantly, subjects who have been cured of Cushing’s disease for a long time still maintain a higher risk for the development of kidney stones compared with controls, which is also associated with a persistence of metabolic syndrome and HTN, despite the normalization of cortisol levels. Hypercalciuria, hypocitraturia, and increased urinary excretion of phosphorus, oxalate, potassium, and cystine, mainly systemic HTN and excess urinary excretion of uric acid, seem to play a crucial role in kidney stone formation in patients with Cushing’s disease [166].
In CS, systemic HTN, which leads to an increased capillary hydrostatic pressure, and additional hemodynamic effects, which cause hyperfiltration, may eventually result in proteinuria and glomerulosclerosis. Glomerulosclerosis has been documented in humans and dogs with CS and in dogs treated with prednisone [183, 189–192].
Diagnosis
One of the largest challenges remains to timely identify patients with CS. In the current society of obese individuals, one may have to consider establishing new reference ranges for various hormones. For instance, norm values for urinary-free cortisol adapted to obese children may help avoid unnecessary dexamethasone suppression or other testing for possible CS [193]. This may even be difficult in full-blown CS, as the sensitivity and specificity of clinical signs and symptoms of CS vary widely. For instance, the occurrence of ecchymoses may have a specificity of 94 % for CS, while its sensitivity varies between 60 and 68 %. Stretch marks wider than 1 cm and of purple color have a sensitivity of 50–64 % and a specificity of 78 %. Increased fatigue and weight gain have a sensitivity of nearly 100 % but are not very specific [194]. This poses the question who should be screened for CS. First, patients with signs that are most suggestive of hypercortisolism, including wide (> 1 cm) purple striae, excessive bruising, proximal muscle weakness, abnormal fat distribution (temporal fossae and supraclavicular), and failure of linear growth with continued weight gain in children. Second, anyone with unusual features for their age group, including HTN and/or nontraumatic fractures in young individuals, or anyone with multiple clinical features that are progressive over time. The Endocrine Society has published guidelines on this topic in 2008 and the algorithm is shown in Fig. 16.6 [195].
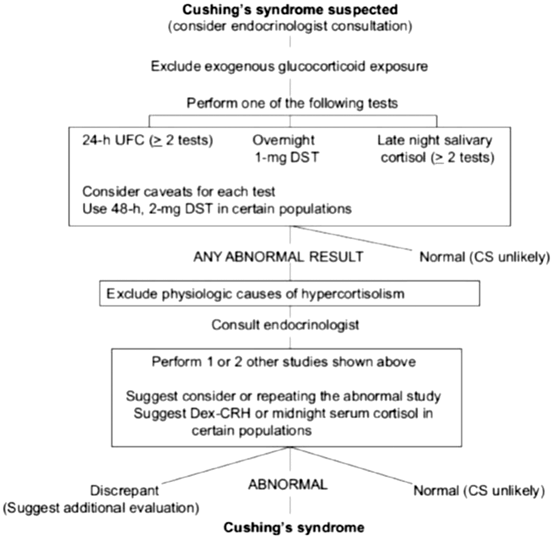
Fig. 16.6
Cushing’s syndrome algorithm. (Reprinted from Nieman et al. [195], with permission from The Endocrine Society)
Biochemical assessment for CS in patients with chronic renal impairment is difficult. Obviously, assessing 24-h urinary-free cortisol excretion largely depends on how precisely an individual patient collects urine and on her/his renal function. Plasma binding protein concentrations and dexamethasone clearance can be significantly altered with decreased renal function [143, 196, 197]. Recent studies have shown a disrupted circadian cortisol rhythm in patients with ESRD , whereas previous studies showed normal rhythms [197–200]. C-reactive protein is increased in patients with ESRD [201]. Some authors report that patients with chronic renal failure generally have normal plasma levels of cortisol, depending on the assay used [202, 203], whereas ACTH levels may be increased [204]. Spuriously, low urinary-free cortisol levels can occur in patients with a GFR less than 30 ml/min [205]. Ovine CRH may not appropriately stimulate ACTH and cortisol in patients with chronic renal failure except for those on continuous ambulatory peritoneal dialysis [206]. The oral absorption of dexamethasone can be altered (reduced) in some patients, but the metabolism of dexamethasone usually is normal in chronic renal failure. Normal suppression of cortisol after 1 mg of overnight dexamethasone is uncommon [207]. The use of the dexamethasone suppression test in the diagnosis of CS has recently been critically reviewed [208]. Even if plasma dexamethasone levels are measured, the interpretation of what a normal cortisol suppression is for each individual patient assessed for possible CS remains challenging, as there are patients with Cushing’s disease with slightly or moderately elevated basal plasma cortisol levels and post dexamethasone plasma cortisol levels that are interpreted as normal [209]. In addition, there are many medications that can alter the metabolism of dexamethasone. Recently, topiramate, a frequently used contemporary medication, has been reported to cause a false positive overnight 1-mg dexamethasone suppression test [210].
Alternatively, salivary cortisol has been regarded as a reliable parameter for the diagnosis of severe hypercortisolism, even in women during pregnancy or taking oral contraceptive pills [211]. Obtaining two samples improves the diagnostic accuracy of measuring late-night salivary cortisol for CS [212]. A study of 16 patients on daytime chronic hemodialysis and controls showed that ESRD subjects have increased late-night plasma and salivary cortisol and plasma ACTH levels, with late-night salivary cortisol being a reliable index of plasma cortisol in ESRD patients [213] . In that study, the authors suggest that ESRD may represent a state of ACTH-dependent hypercortisolism but not CS per se, similar to previous studies in poorly controlled diabetics [214]. Raff and Trivedi [213] measured late-night salivary cortisol levels as high as 15 nmol/l in ESRD patients (reference range, < 4 nmol/l or 145 ng/dl, ref. [195]) and concluded that a “normal” late-night salivary cortisol value rules out CS in patients with ESRD.
Interestingly, ACTH-stimulated salivary cortisol represents an accurate biomarker for the diagnosis of adrenal insufficiency in hypotensive patients with ESRD, whereas neither basal salivary nor serum cortisol do [215].
Genetic Aspects of Cushing’s Syndrome
We here will not review the molecular pathogenesis of ACTH-secreting pituitary tumors, ectopic ACTH-secreting neuroendocrine tumors, or adrenal tumors. However, a few statements based on more recent findings should be made. In general, the molecular pathogenesis of ACTH-independent macronodular adrenal hyperplasia and sporadic cortisol-secreting adenomas has recently been elucidated. Exome sequencing or tumor–normal pairs revealed gain of function mutations in the CTNNB1 (β-catenin) or GNAS (Galphas) genes or somatic mutations in the PRKACA (protein kinase A catalytic subunit) gene with protein kinase activity inducing cortisol production and cell proliferation of affected adrenal cells [216]. Germline duplications of the PRKACA gene result in bilateral adrenal hyperplasia, whereas somatic mutations in this gene cause unilateral cortisol-producing adrenal adenomas [217]. A subset of ACTH-independent macronodular adrenal hyperplasia contains inactivating mutations in armadillo repeat containing 5 (ARMC5), a putative tumor-suppressor gene [218]. Regarding ACTH in the pituitary, most of such tumors occur sporadically with altered gene expression, somatic mutations in various genes, epigenetic changes, and abnormal microRNAs. Their pathogenesis remains largely unknown [219]. Whole exome sequencing of pituitary adenomas reveals different oncogenic mutations in each individual tumor, making it probable that there is no common oncogenic denominator but an abnormal stem cell that leads to abnormal localized proliferation. Nevertheless, there are familial pituitary tumor syndromes, including multiple endocrine neoplasia type 1, Carney complex, familial isolated pituitary adenomas, and others. The pathogenesis of ectopic ACTH-secreting neuroendocrine tumors is largely unknown, although some familial syndromes in which an ACTH-secreting neuroendocrine tumor is part of helped elucidate the biology of these neoplasms. For instance, neuroendocrine tumors of the pancreas in patients with VHL syndrome usually are nonfunctional but can secrete ACTH [220]. VHL syndrome is caused by germline mutations in the VHL gene and subsequent molecular events [221]. Neuroendocrine tumors of the thyroid usually are medullary thyroid cancer and often are caused by germline mutations in the rearranged during transfection (RET) proto-oncogene [222]. Many other organs can harbor ACTH-secreting neuroendocrine tumors, and for each organ, for instance, the prostate, pancreas, adrenal medulla/pheochromocytoma, one would have to consider the respective pathogenesis [223–225].
Therapy
In patients with a cortisol-producing adrenal tumor, laparoscopic adrenalectomy should be performed to normalize cortisol production. If bilateral adrenalectomy is necessary, proper adrenal hormone replacement should be commenced. For instance, most individuals can be managed by taking oral hydrocortisone, 10 mg in the morning and 5–10 mg late afternoon, in addition to fludrocortisone 50–100 mcg/day [226]. Adrenal function should be assessed in patients with ESRD in order to unmask adrenal insufficient states [227].
In patients with an ACTH-secreting pituitary tumor, transsphenoidal or transcranial surgical removal should be performed, if possible. Postsurgically, transient or permanent adrenal insufficiency can occur and should be handled, as aforementioned. Long-term follow-up is necessary even in patients with initial and long-term remission [228]. In patients with nonpituitary ACTH-dependent CS, ideally the primary neuroendocrine tumor should be found and removed, although surgical resection of metastases might result in short-term remission of CS [60]. For patients who are not or poor surgical candidates, medical therapy of hypercortisolism should be initiated. Depending on country-specific regulations and availability of drugs, one can choose among the following arsenal with each one to be revisited for mild or more impaired renal and/or liver function with respective dose adjustments: metyrapone, ketoconazole, mifepristone RU486, cabergoline, pasireotide, octreotide, etomidate, and mitotane [195, 229–232].
Related posts:
 Neurogenic Factors in Hypertension Associated With Chronic Kidney Disease
Dual Inhibitors: RAAS Blockers/Combination Therapies: What Do All These Trials Mean?
Genome-Wide Association Studies (Gwas) of Blood Pressure in Different Populations
Neurogenic Factors in Hypertension Associated With Chronic Kidney Disease
Dual Inhibitors: RAAS Blockers/Combination Therapies: What Do All These Trials Mean?
Genome-Wide Association Studies (Gwas) of Blood Pressure in Different Populations
 Resistant Hypertension in Patients with Chronic Kidney Disease
Resistant Hypertension in Patients with Chronic Kidney Disease
 Hypertension in Children with Chronic Kidney Disease
Hypertension in Children with Chronic Kidney Disease
 Changes in Guideline Trends and Applications in Practice: JNC 2013 and the Future
Changes in Guideline Trends and Applications in Practice: JNC 2013 and the Future
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


