Infection with hepatitis C virus (HCV) is a worldwide epidemic affecting up to 3% of the world’s population. Approximately 80% of people infected with HCV will go on to develop chronic disease. Of these individuals, approximately 25% develop cirrhosis and are vulnerable to its complications, including hepatocellular carcinoma. Treatment for HCV can be curative, and successful treatment improves the quality of life of HCV infected individuals as well as prevents progression of liver disease and its associated morbidity and mortality.
For the last decade, the standard of care treatment for HCV has been pegylated interferon-α (pegIFNα) plus ribavirin (RBV) for 24 to 48 weeks depending on viral genotype. Therapy is expensive, difficult to tolerate, and associated with a variety of quality-of-life–limiting side effects. Furthermore, the response rates to treatment are not optimal; only about 40% of genotype 1–infected patients respond to treatment. Patients infected with genotypes 2 and 3 have a higher rate of treatment response, which approximates 80%.
A major goal within the fields of hepatology and infectious diseases is to improve the rate of response to HCV treatment. Novel discoveries over the past few years, such as the finding of a polymorphism near the IL28B gene, have helped identify those patients that are most likely to respond to HCV treatment with pegIFNα and RBV. Additionally, new therapies have been developed, and some of the most promising new drugs are those that act to directly inhibit the virus. These direct-acting antiviral (DAA) drugs are currently in various stages of development, and are described in detail in this review.
Viral Structure and Life Cycle
A basic understanding of the viral structure and life cycle ( Fig. 1 ) is important to understand the targets of the new drugs in development. The HCV is a positive single-strand RNA virus with 9.5 kilobases. The virus enters the hepatocyte via a variety of receptors (including glycosaminoglycans, CD81, SR-BI, Claudin-1) by receptor mediated endocytosis. Once in the hepatocyte, the virus is translated into a single long polypeptide on the ribosome then cleaved by both host and viral proteases into 10 functional proteins. A replication complex is formed using viral and host proteins and results in a double-stranded RNA intermediate that includes a positive-strand RNA and a negative-strand RNA. The negative strand serves as a template for the synthesis of positive-strand RNA, which is then packaged and released from the hepatocyte. The synthesis of positive-strand RNA is disproportionate to the negative strand and is transcribed in a 5- to 10-fold excess of negative-strand RNA.
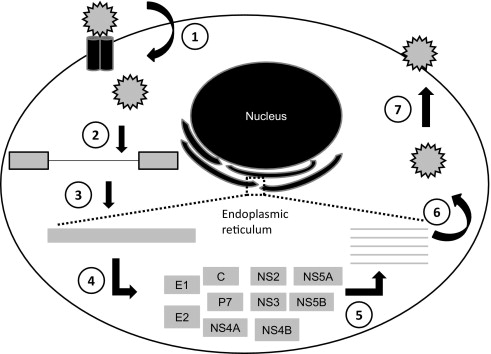
HCV is fairly unique in that there is not a DNA intermediate in the life cycle. Thus, the virus does not incorporate itself into the host DNA. The result is the ability to effectively cure the infection indefinitely. This is in contrast with the hepatitis B virus or HIV, where suppression is the major goal of therapy.
The viral RNA produces 10 functional proteins. These proteins are composed of structural and nonstructural proteins. The nonstructural proteins are involved in viral replication and processing and are the target of most DAAs. These are described in more detail herein.
Viral Resistance
An important barrier in the development of antiviral agents is the emergence of virus resistance. There are several qualities of HCV that make it particularly prone to developing drug-resistant strains. The virus has a high rate of replication with 10 12 virions produced daily. The viral protein that mediates viral replication is NS5B, or RNA-dependent RNA-polymerase. This protein lacks proofreading ability and has a high error rate, thus leading to a variety of genetic strains of virus. In general, viruses that are mutated versions of the wild type have less replication fitness than wild-type virus, and so are present in the blood at lower quantities. Some of these mutated viruses result in a change to the viral protein structure; thus, if this occurs at the site where a DAA acts, it may not be effective in inhibiting the action of the protein. Often these drug-resistant strains of virus are present at low levels in the blood at baseline, but when subjected to selection pressure, such as the addition of a DAA, the wild-type virus decreases and the mutated virus increases and ultimately renders the DAA ineffective ( Fig. 2 ).
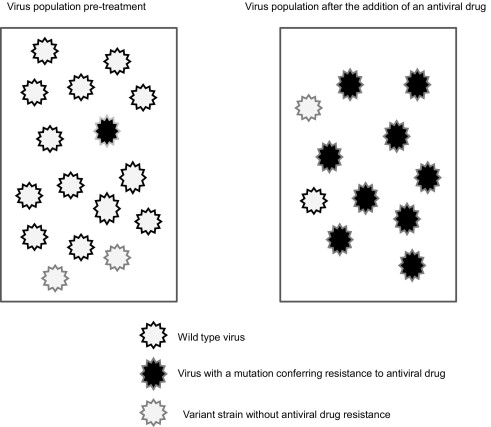
Viral Resistance
An important barrier in the development of antiviral agents is the emergence of virus resistance. There are several qualities of HCV that make it particularly prone to developing drug-resistant strains. The virus has a high rate of replication with 10 12 virions produced daily. The viral protein that mediates viral replication is NS5B, or RNA-dependent RNA-polymerase. This protein lacks proofreading ability and has a high error rate, thus leading to a variety of genetic strains of virus. In general, viruses that are mutated versions of the wild type have less replication fitness than wild-type virus, and so are present in the blood at lower quantities. Some of these mutated viruses result in a change to the viral protein structure; thus, if this occurs at the site where a DAA acts, it may not be effective in inhibiting the action of the protein. Often these drug-resistant strains of virus are present at low levels in the blood at baseline, but when subjected to selection pressure, such as the addition of a DAA, the wild-type virus decreases and the mutated virus increases and ultimately renders the DAA ineffective ( Fig. 2 ).
NS3/4A Protease Inhibitors
The NS3/4A protease acts to cleave the polyprotein into its various functional proteins. First-generation agents that target this protease—boceprevir and telaprevir—are the furthest along in development. Several phase II and III study results are available and are summarized. The primary end point of HCV clinical trials is achievement of sustained virologic response (SVR), which is absence of HCV RNA in the blood 6 months after the completion of treatment.
Boceprevir
Early phase studies of boceprevir revealed that viral load reductions were greater, and the development of resistance lower, when boceprevir was given in combination with pegIFNα. Subsequent early phase studies showed that the highest rates of viral load reductions were seen in patients who received boceprevir 800 mg 3 times daily in combination with both pegIFNα and RBV and served as the basis for the study designs of phase II and III studies. Boceprevir trials have largely used a lead in phase of 4 weeks of pegIFNα and RBV followed by varying courses of triple therapy with boceprevir, pegIFNα, and RBV.
Boceprevir phase II studies
The goal of the Serine Protease Inhibitor Therapy (SPRINT)-1 trial was to determine the effectiveness of boceprevir in combination with pegIFNα and RBV in genotype 1 treatment-naïve patients, and to determine whether a lead in phase of pegIFNα/RBV for weeks before boceprevir therapy improved response rates. A second part of the study evaluated whether lower dose RBV is as effective as standard dose RBV.
Table 1 details the treatment groups and results, including achievement of SVR and relapse. The highest rates of SVR with the lowest relapse rates were seen in the groups that received a total of 48 weeks of therapy (SVR 75% and 67% in lead in group and 48 week treatment group, respectively). The groups that received a total of 28 weeks of therapy had better rates of SVR than the control group (SVR 54% and 56% vs 38%), but had relatively high relapse rates (30% and 24%). The control group had no viral breakthrough, defined as a persistent 2-log 10 or greater increase from nadir and 50,000 IU/mL or higher. The lead-in groups had less viral breakthrough than the groups with no lead in (4% vs 9%), although this did not reach significance ( P = .057). Phase III studies were designed using a lead in strategy for all boceprevir groups to minimize viral breakthrough.
| Study/Treatment Group | SVR (%) | Relapse (%) | |||
|---|---|---|---|---|---|
| SPRINT-1 | |||||
| B/P/R 28w | 54 | 30 | |||
| B/P/R 48w | 67 | 7 | |||
| P/R 4w, B/P/R 24w | 56 | 24 | |||
| P/R 4w, B/P/R 44w | 75 | 3 | |||
| P/R 48w | 38 | 24 | |||
| B/P/R 48w – Standard dose R | 67 | 7 | |||
| B/P/R 48w – Low dose R | 36 | 22 | |||
| Nonblack | Black | Nonblack | Black | ||
|---|---|---|---|---|---|
| SPRINT-2 | |||||
| P/R 4w, RGT | 67 | 42 | 23 | 14 | |
| P/R 4w, B/P/R 44w | 68 | 53 | 8 | 17 | |
| P/R 48w | 40 | 23 | 23 | 14 |
| Overall | Prior Relapse | Prior Partial Response | Overall | ||
|---|---|---|---|---|---|
| RESPOND-2 | |||||
| P/R 4w, RGT | 59 | 69 | 40 | 32 | |
| P/R 4w, B/P/R 44w | 66 | 75 | 52 | 12 | |
| P/R 48w | 21 | 29 | 7 | 32 | |
In the second part of the trial, a “low-dose” RBV group (400–1000 mg) was compared with a “standard dose” RBV group (800–1400 mg). The low-dose RBV group had lower SVR (36% vs 67%) and higher relapse (22% vs 7%) than the standard dose RBV group. Rates of viral breakthrough were high in both of these groups (27% in low dose and 25% in standard dose). The results of SPRINT-1 suggest that low-dose RBV is not an adequate treatment option.
Boceprevir phase III studies
Table 1 details the treatment groups studied in phase III trials and summarizes the results. The goal of SPRINT-2 was to evaluate the effectiveness of response-guided treatment (RGT) with boceprevir in genotype 1, treatment-naïve patients in 2 cohorts: Non-black patients and black patients. In the RGT group, patients who achieved undetectable HCV RNA by week 8 and remained negative for HCV RNA at week 24 completed 28 weeks of therapy. Those who did not achieve undetectable HCV RNA by week 8 received a total of 48 weeks of therapy. This group was compared with a group that received the full 48 weeks of therapy. All patients received a 4-week lead-in period with pegIFNα and RBV alone.
The RGT group had similar SVR rates as the group that received 44 weeks of boceprevir (67% vs 68% in the non-black cohort and 42% vs 53% in the black cohort). All boceprevir groups had significantly higher SVR rates than the standard therapy groups. Of the nonblack patients in the RGT group, 47% were eligible to receive a shortened course of therapy (28 weeks). The patients who received 28 weeks of therapy had an SVR rate of 97% and the patients who received 48 weeks of therapy had an SVR rate of 98%. Thus, SPRINT 2 provided the data to support 28 weeks of combination therapy in patients that achieve undetectable viral loads at weeks 8 through 24.
The goal of RESPOND-2 was to assess the response of boceprevir therapy in genotype 1 patients who had prior relapse (undetectable HCV RNA at the end of prior therapy without subsequent attainment of SVR) or partial response (decrease in HCV RNA level of ≥2 log 10 IU/mL by week 12 of prior therapy but detectable HCV RNA level throughout the course of therapy, without achievement of SVR) to standard treatment. Patients with prior null response (lack of 2-log 10 reduction in HCV RNA at week 12) were excluded from this study. Patient groups were similar to those in SPRINT-2 and included an RGT group in which patients who achieved a negative viral load at week 8 received only 36 weeks of therapy. The RGT group and the group that received 48 weeks of triple therapy had similar results and were both superior to the control group (SVR 59% and 66% vs 21%). Better response rates were seen in patients with prior relapse to treatment than prior partial response. In the RGT group, 46% of patients were eligible to receive a shorter course of therapy, and these patients had similar rates of SVR when they received a total of 36 weeks of treatment compared with those that received a total of 48 weeks of treatment. Thus, RESPOND-2 supports the use of a shorter course of treatment (36 weeks) for patients with prior relapse and partial response if they achieve a negative viral load at week 8 of treatment.
Adverse events with boceprevir
In general, boceprevir is well-tolerated. However, there is a higher rate of treatment discontinuation and dose reductions in patients that receive boceprevir in combination with pegIFNα and RBV than those that receive standard therapy alone (treatment discontinuation 8%–16% vs 3%–16% and dose reduction 29%–40% vs 14%–26%). The most common side effects experienced by patients are typical pegIFNα and RBV related side effects such as flu-like symptoms, fatigue, and nausea. Effects that are more prevalent in those who receive boceprevir are anemia and dysgeusia. Anemia developed in 43% to 56% of patients who received boceprevir in combination with pegIFNα and RBV in phase II and III trials. In SPRINT-2, erythropoietin was administered in 24% of the controls and 43% of boceprevir recipients. Another side effect with boceprevir combination therapy is dysgeusia, which occurred in 37% to 43% of patients.
Telaprevir
Telaprevir monotherapy was also determined to have high rates of viral load reduction, but also resistance development in early phase studies; therefore, its development proceeded with concurrent pegIFNα and RBV. The treatment regimens with telaprevir differ from those described for boceprevir. Early phase studies supported using combination therapy for the first 12 weeks followed by pegIFNα and RBV for 12 to 36 additional weeks. This is opposed to boceprevir, where a lead-in with pegIFNα and RBV was followed by use of the combination therapy. The dose of telaprevir studied in phase II and III trials was 750 mg every 8 hours.
Phase II studies
Multiple phase II studies have been reported with telaprevir, including Protease inhibition for Viral Evaluation (PROVE)-1, -2, and -3. Table 2 details the treatment groups and results for telaprevir phase II studies.
Related posts:
 Noninvasive Tools to Assess Hepatic Fibrosis: Ready for Prime Time?
Noninvasive Tools to Assess Hepatic Fibrosis: Ready for Prime Time?
 Acute Liver Failure: Current Practice and Recent Advances
Long-Term Management of the Liver Transplant Recipient: Pearls for the Practicing Gastroenterologist
Noninvasive Tools to Assess Hepatic Fibrosis: Ready for Prime Time?
Liver Transplantation in the 21st Century: Expanding the Donor Options
Alcoholic Hepatitis: Prognostic Models and Treatment
Acute Liver Failure: Current Practice and Recent Advances
Long-Term Management of the Liver Transplant Recipient: Pearls for the Practicing Gastroenterologist
Noninvasive Tools to Assess Hepatic Fibrosis: Ready for Prime Time?
Liver Transplantation in the 21st Century: Expanding the Donor Options
Alcoholic Hepatitis: Prognostic Models and Treatment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


