FIGURE 6.1 Internal and external potassium balance. The figure depicts typical potassium balance in a 70-kg man with a total body water of 42 L.
■ Distal tubule—changes in urinary potassium excretion usually reflect the amount of potassium secretion by this nephron segment—<10% of filtered potassium reaches the distal convoluted tubule, but fraction of filtered K+ leaving late distal tubule is 20%, indicating active secretion into distal tubule
■ Cortical collecting tubule—secretes K+ in response to active Na+ uptake (site of aldosterone action)
■ Medullary collecting tubule and duct—in response to K+ excess, secretion of K+ into the lumen can occur—usually no net K+ flux
FACTORS INFLUENCING RENAL POTASSIUM EXCRETION
■ Mineralocorticoid hormone (aldosterone)—enhances active transport of K+ from blood to distal tubular cell and increases Na+ permeability of luminal membrane, thus increasing K+ secretion
■ Acid–base balance—acute metabolic acidosis decreases intracellular K+ and K+ excretion; acute metabolic alkalosis increases intracellular K+ and K+ excretion; however, both chronic metabolic acidosis and alkalosis are associated with increased K+ excretion (Adrogué & Madias, 1981)
■ Anion effects—nonchloride anions stimulate K+ secretion
■ Tubular flow rate—increases in urinary flow increase K+ excretion independent of sodium delivery—antidiuretic hormone increases K+ excretion; this effect helps to maintain external K+ balance under conditions of antidiuresis
■ Sodium intake—increased Na+ intake increases K+ excretion; decreased Na+ intake decreases K+ excretion
■ Potassium intake—K+ loading causes decreased reabsorption by TAL and increased K+ secretion by distal nephron segments
■ Intracellular and plasma potassium concentration—increases in plasma (extracellular) [K+] result in increased K+ excretion
■ Diuretics—osmotic diuretics (urea, glucose, mannitol), thiazides, carbonic anhydrase inhibitors, loop diuretics all increase K+ excretion by increasing fluid delivery to distal tubule; loop diuretics also block K+ reabsorption by TAL. Spironolactone (aldosterone antagonist), triamterene, and amiloride all decrease K+ excretion at the level of distal tubule
■ Magnesium—A decrease in intracellular magnesium, caused by magnesium deficiency, decreases the magnesium-mediated inhibition of renal outer medullary potassium (ROMK) channels and increases potassium secretion
GASTROINTESTINAL HANDLING OF POTASSIUM
■ No role in normal potassium homeostasis
■ In chronic renal failure and potassium loading, fecal excretion of K+ increases several-fold due to active colonic K+ secretion, at least part of which is aldosterone dependent
CONTROL OF INTERNAL POTASSIUM BALANCE
■ Insulin—physiologic regulator of plasma [K+]—stimulates uptake of K+ into cells (primarily muscle and liver)—effect mediated by Na+–K+ pump and can be blocked by ouabain
■ Clinical significance: Insulinopenia is associated with hyperkalemia (e.g., diabetic ketoacidosis); insulin is useful in emergency treatment of hyperkalemia
■ Catecholamines—α-receptor stimulation increases plasma K+; β2 agonists decrease plasma K+ (no effect of β1 agonists)
■ Clinical significance: conditions associated with increased circulating catecholamines (myocardial ischemia, pheochromocytoma) associated with hypokalemia; β2 agonists can be used to treat hyperkalemia
■ Hypertonicity—infusion of hypertonic solutions results in water and K+ movement out of cells
■ Clinical significance: uncertain, though hypertonic glucose (given for diabetic coma) may result in hyperkalemia
■ Acid–base balance—K+ distribution affected by pH, with acidosis causing K+ shift out of cells and alkalosis shifting K+ into cells (Adrogué & Madias, 1981). However, relationship between acidemia and hyperkalemia complex—in inorganic metabolic acidosis (e.g., renal failure, HCl administration, H+ –K+ exchange leads to hyperkalemia, whereas in organic metabolic acidosis (e.g., lactic acidosis, ketoacidosis), when H+ ions move into cells, they are accompanied by organic anions. Since electroneutrality is maintained, no need for K+ exit from cells; thus, little or no increase in plasma [K+]
■ Clinical significance: hyperkalemia associated with organic acidosis probably not due to acidosis per se but secondary to associated events (e.g., tissue breakdown, renal failure, insulinopenia)
HYPOKALEMIA
Etiologies of Hypokalemia
■ Hypokalemia due to transcellular redistribution (increased K+ uptake into cells with total body potassium usually normal)
■ Alkalemia—increased H+ exit and K+ uptake by cells
■ Periodic paralysis—Weakness during hypokalemia that usually lasts 3 to 4 hours but may last up to 24 hours
• Hereditary form—attacks may be precipitated by high carbohydrate meals (increased insulin release) or exercise (increased circulating catecholamines) (voltage-gated channel gene mutation)
• Acquired form—associated with thyrotoxicosis (thyroid-hormone sensitive K+ transporter mutation). Resolves with treatment of thyrotoxicosis
• Administration of insulin or β2 adrenergic agonists (or increased endogenous secretion)—shift K+ into cells
■ Hypokalemia with low total body potassium
■ Decreased intake—inadequate K+ in diet (e.g., in elderly and alcoholic patients)
■ Gastrointestinal potassium losses—vomiting, diarrhea, laxative abuse, villous adenoma
■ Renal potassium losses
• Associated with hypertension—Conn syndrome (primary mineralocorticoid excess) (Conn et al., 1985; Blumenfeld et al., 1994), renovascular hypertension (secondary mineralocorticoid excess), Cushing disease or iatrogenic steroids (glucocorticoid excess), licorice abuse (increases mineralocorticoid effect of cortisol), diuretics (increase fluid delivery to distal tubule)
• Associated with normal blood pressure—Renal tubular acidosis (covered in acid/base chapter), osmotic diuresis, diuretics, antibiotics (increased anion delivery to distal tubule), Bartter syndrome (defect in ion transport in the loop of Henle), Gitelman syndrome (defect in ion transport at distal tubule) (Gladziwa et al., 1995), leukemia (associated with lysozymuria), magnesium depletion (increases renal potassium excretion) (Whang et al., 1985)
• In the setting of increased distal sodium delivery or elevated aldosterone, depletion of magnesium can cause potassium secretion
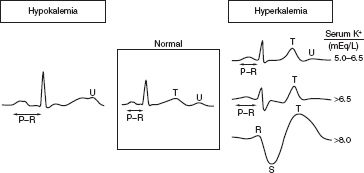
FIGURE 6.2 EKG changes in hypo- and hyperkalemia.
Consequences of Hypokalemia
■ Cardiovascular effects: Electrocardiographic (EKG) changes (U waves) (Fig. 6.2)
■ Neuromuscular effects: weakness (even quadriparesis), rhabdomyolysis, ileus, encephalopathy (in patients with liver disease)
■ Metabolic and hormonal effects: abnormal carbohydrate metabolism (decreased insulin release and peripheral effect), negative nitrogen balance, decreased aldosterone release
■ Renal effects: polyuria, polydipsia (nephrogenic diabetes insipidus), increased renal NH3 production (can cause hepatic encephalopathy)
Laboratory Evaluation of Hypokalemia
■ Urinary K <20 mmol/L = extrarenal loss
■ Urinary K >20 mmol/L = renal loss
■ Urinary Cl <10 mmol/L + urinary K >20 mmol/L = vomiting (HCl loss)
■ Transtubular potassium gradient (TTKG) = [(Urine K+ / Plasma K+) / (Urine osm / Plasma osm)] (Fig. 6.3) (Joo et al., 2000)
■ In hypokalemic periodic paralysis and extrarenal loss, TTKG is low (~2)
■ In hyperaldosteronism, TTKG is high (>5)
■ For proper interpretation, urine [Na+] >25 mmol/L; urine osm > plasma osm
Symptoms of Hypokalemia
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


