Glucose-6-phosphatase
Adrenal insufficiency
Familial hypoparathyroidism
Nephrogenic diabetes insipidus
Mauriac’s syndrome
Familial cholestatic syndrome
Anorexia nervosa
Severe malnutrition
Atopic dermatitis
Keratitis–ichthyosis–deafness (KID) syndrome
Fucosidosis
Pseudohypoaldosteronism
Patients undergoing prostaglandin infusions
Hyperchlorhidrosis caused by homozygous mutation in CA12, encoding carbonic anhydrase XII. [25]
Accurate references lacking
Glucose-6-phosphate-1-dehydrogenase deficiency
Ectodermal dyplasia
Hypothyroidism
At the other end of the spectrum, problems arise when there is a CF phenotype in one or more organ systems, and the sweat chloride level is borderline (30–60 mmol/L) .
Electrophysiological testing using nasal potential difference (NPD) measurements may aid in the diagnosis of CF patients with normal or borderline sweat tests and negative or uninformative genetic test results [26]. This test measures transepithelial sodium and chloride transport in the nasal epithelium. The function of CFTR in chloride secretion is intimately related to the inwardly directed sodium transport via the epithelial sodium channel (ENaC). The activity of these two channels is the basis for NPD. A catheter is placed under the inferior turbinate and is connected via a series of electrodes to a voltmeter and recorder. The nasal epithelium is perfused with solutions which inhibit sodium transport and activate chloride transport. In CF patients, the readings are markedly dissimilar from controls. Another electrophysiological tool is the intestinal current measurement (ICM) which measures CFTR function ex vivo in rectal biopsy using a modified Ussing chamber [27]. Testing should only be carried out by an experienced individual in specialist research center where objective reference values have been established.
CFTR Dysfunction: Gastrointestinal Consequences
Exocrine Pancreatic Abnormalities
Exocrine Pancreatic Function
In pancreatic ductal epithelia, the CFTR protein is highly expressed, allowing fluids and anion to enter the ductal lumen. Thereby, luminal chloride is exchanged for bicarbonate, giving evidence that CFTR is permeable to bicarbonate [28]. According to the Quinton’s hypothesis, the defect in bicarbonate transport is indeed the primary defect in CF [29]. This results in an increased volume of alkaline fluid, allowing the acinar cells to remain in a soluble state, due to the highly concentrated proteins secreted. Absent or reduced CFTR channel function impairs chloride and bicarbonate to enter the ducts which results in reduced volume of a more acidic fluid [30]. The acidic milieu within the acinar lumen also leads to impaired reuptake of GP2, the zymogen-granule-associated protein [31]. The consequences of mutations in the CFTR gene have been demonstrated by pancreatic function studies showing that CF patients have low flow secretions with a high protein concentration, presumably which will precipitate in the duct lumina causing obstruction, damage, and atrophy (Fig. 41.1).
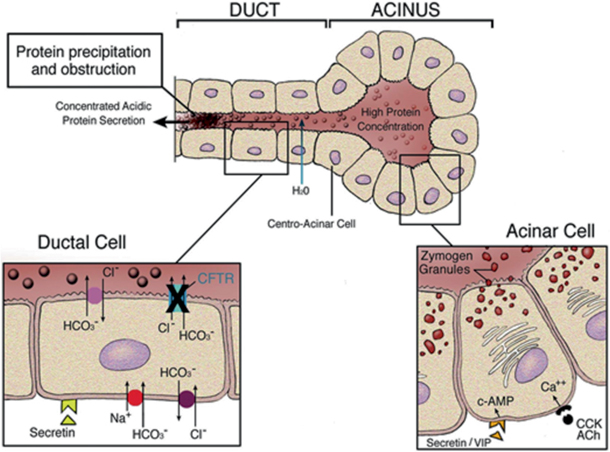
Fig. 41.1
Pathogenesis of pancreatic disease in cystic fibrosis (CF) [32]. Acinar cells secrete large quantities of protein, primarily in the form of digestive enzymes, into the acinar lumen. Under normal circumstances, anions (chloride and bicarbonate) are secreted into the ductal lumen via the cystic fibrosis transmembrane regulator (CFTR) and bicarbonate exchangers. This provides a driving force for the movement of fluid into the lumen of the duct and maintains the solubility of secreted proteins in a dilute, alkaline solution. In CF, impaired anion transport into the proximal ducts results in decreased secretion of more acidic fluid, which leads to precipitation of secreted proteins. Intraluminal obstruction of the ducts then causes progressive pancreatic damage and atrophy. (Reproduced from Ref. [32], Copyright 2007, with permission from BMJ Publishing Group Ltd) cAMP cyclic adenosine monophosphate, ACh acetylcholine, CCK cholecystokinin, VIP vasoactive intestinal peptide
These changes begin in utero, and after birth, the process continues with the small duct obstruction leading to a larger duct obstruction. For several months afterwards, there is a release of proteins, originating in the pancreas, into the blood stream. This pathological process forms the basis for the immune reactive trypsinogen (IRT), the neonatal screening test for CF. The reason has yet to be determined, but, interestingly enough, the infant is asymptomatic while this wholesale destruction of the exocrine pancreas is occurring. Eventually, this process results in severe inflammatory changes, obstruction of ducts by mucus and calcium-containing debris, the destruction of acini, and generalized fibrosis. The high IRT does show that some exocrine pancreatic tissue is still present and may have a bearing on possible small molecule therapy targeted at the remainder of the pancreas which may rescue enough tissue to cause viability of the remaining pancreas. This of course contradicts the popular belief that the pancreas is entirely nonfunctioning at birth.
One of the most remarkable observations is that genetic factors exquisitely influence the degree of pancreatic disease and its rate of progression. CF patients are classified as pancreatic insufficient (PI) or pancreatic sufficient (PS). PI patients comprise approximately 85 % of all CF patients. PI patients present with maldigestion, as evidenced by steatorrhea (fecal fat greater than 7 % of fat intake in infants over 6 months of age and over 15 % under 6 months of age), requiring these patients to have pancreatic enzyme replacement therapy (PERT) with meals. PS patients, however, have evidence of pancreatic damage, but retain sufficient endogenous exocrine pancreatic function to sustain normal digestion [33] due to the fact that ductal CFTR in PS patients is partially functional, thereby allowing anions and fluid to enter the ductal lumen. This provides further evidence that mutant CFTR may be acting on other apical anion exchangers to conduct rather than conduct chloride ions itself [34].
The exocrine pancreatic status is directly linked to genotype [35]. Analysis of particular CFTR mutations in patients with pancreatic phenotypes (PI vs. PS) revealed two categories of alleles: “severe” and “mild.” Patients homozygous or compound heterozygous for severe alleles belonging to classes I, II, III, or VI confer PI as opposed to patients with a mild class IV or V allele even when the second mutation is severe. These patients are termed “PS.” Plausible explanation of this observation finds all known mild alleles belong to class IV or class V all of which are (or predicted to be) associated with some residual chloride channel activity at the epithelial apical membranes. Although these mutations confer sufficient CFTR function to prevent the pancreas to be completely destroyed, many PS patients have reduced exocrine pancreatic capacity and are associated with an increased risk of pancreatitis, as discussed below. A very small number (2–3 %) of patients carrying severe mutations on both alleles are PS at diagnosis; however, most patients will experience a gradual transition from PS to PI. A few missense mutations (e.g., G85E) confer a variable pancreatic phenotype.
Many PS patients have reduced exocrine pancreatic capacity and are associated with an increased risk of pancreatitis, even though mild mutations confer sufficient CFTR function to prevent the pancreas to be completely destroyed. As first reported by Shwachman in 1975, recurrent acute and chronic pancreatitis are relatively infrequent complications of CF. In this retrospective study, only 0.5 % of CF patients had pancreatitis. Durno et al. reported more recently, an incidence of 1.7 % in a cohort of over 1000 patients followed over a period of 30 years [36]. All of the pancreatitis patients were classified as PS, but this subgroup of PS patients, in fact, appeared to be highly susceptible to pancreatitis, since almost one in five was affected by this complication. In a seminal paper of the largest study to date of CF PS patients, Ooi et al. determined the association between severity of CFTR genotype and the risk of pancreatitis [37]. They examined a large cohort of 277 PS patients from 2 CF centers of which 62 had well-documented pancreatitis. The mutations were divided into three main groups: severe, moderate–severe, and mild in using a novel pancreatic insufficiency prevalence score. It was found that the proportion of patients who developed pancreatitis was significantly greater for genotypes in the mild group than the moderate–severe group. Thus, the more mild mutations were associated with increased risk of pancreatitis. CFTR mutations may contribute to the development of pancreatitis along with other genetic and environmental factors [38].
Diagnosis of Pancreatic Phenotype
With the increasing worldwide use of neonatal screening programs, patients may present with symptoms of CF or be entirely asymptomatic. Making a determination in all patients, whether the patient is PI or PS is essential to enable a rational use of oral PERT [39]. When patients have steatorrhea (oil droplets on stool microscopy), hypoalbuminemia , and low fat-soluble vitamin levels, the diagnosis of PI is straightforward. However, the lack of these findings does not exclude PI; therefore, more formal testing is required. In the past, direct exocrine pancreatic stimulation testing was administered. Unfortunately, this was an invasive, time-consuming procedure and not widely used in most centers. Indirect pancreatic testing with 72 h fecal fat measurements should be encouraged and be used as follow up for pancreatic exocrine status and response to therapy. Due to the technical difficulties surrounding the performance of this test, however, most laboratories are utilizing the fecal elastase-1 test instead, as the main advantage is no need for prolonged stool collection. Pancreatic elastase is secreted into the duodenum and is found at relatively high concentrations in stool (> 500 µg/gm stool).
The use of fecal pancreatic elastase-1 has now become a common diagnostic test for assessing exocrine pancreatic status. The advantages and limitations of fecal elastase-1 in CF have been discussed by Kalnins et al. in a review article [40]. The cutoff levels of fecal elastase for PI range between 100 and 200 µg/g stool; a majority of centers use the upper level of 200 µg/g stool. With the use of 200 µg/g stool weight, some patients may be falsely identified as PI. The Toronto group compared stool elastase values to the 72-h fecal fat in both known PI and sufficient patients and found that an elastase value of 100 µg/g stool had a 99 % predictive value in ruling out PI based on an abnormal fecal fat. Cade et al. showed that patients with pancreatic sufficiency on the basis of a normal fecal fat balance study were found to have fecal elastase values in the range of 100–200 µg/g stool; elastase levels were compared with the fecal fat test coefficient of fat absorption (CFA) [41]. Whether defining PI as < 93 or 90 % CFA, cutoff levels of fecal elastase of < 100 µg or < 200 µg/g stool for either monoclonal or polyclonal methods were positive predictors of insufficient pancreatic function. However, patients with pancreatic sufficiency were not included in this study population. Moreover, these observations question the validity of defining the cutoff for PI as a fecal elastase value below 200 µg/g stool. In a different study of 21 known PS patients, a poor correlation was found between fecal fat excretion and elastase-1 [42]. The fecal elastase result will define clearly whether a patient is PI or sufficient, in a majority of cases, provided the test is done accurately. In situations such as in acute diarrhea, short gut, or stool from an ileostomy, where stool is more watery, fecal elastase levels may be lower than expected; therefore, it would be advisable to wait until the diarrhea resolves, or until a sample that is more formed is available. A definitive answer on pancreatic status may not be provided in those patients with less common genetic variations, where there are no supportive clinical features of PI , and fecal elastase values are often borderline. In such cases, elastase can be used to monitor pancreatic status, in conjunction with ongoing evaluation of clinical and nutritional status [43].
Oral Pancreatic Enzyme Replacement Therapy
Prior to 2010, pancreatic enzyme products were exempt from the Food, Drug, and Cosmetic Act of 1938 and did not require approval of the Food and Drug Administration (FDA). Since then, a plethora of products became available to the public without the need for strict preclinical and clinical studies. The FDA has since mandated that all manufacturers of pancreatic enzyme products in the USA must seek approval by April 2010.
Currently, there are six preparations approved and available for use [44–46]: Creon [47–50], Zenpep [51], Pancreaze, Ultresa, Viokace, and Pertzye. During the study on Creon, a fixed dose of 72,000 lipase units with meals and 36,000 with snacks was evaluated. The study was a double-blind, randomized, placebo-controlled trial of 54 adult patients with chronic pancreatitis or post-pancreatic surgery. The treatment arm showed difference in CFA of 19.3 % over placebo (85.6 vs. 66.3).
Creon and Zenpep showed a similar nutrient absorption rate between 83 and 87 % fat absorption. The dosages of these products are based on the lipase units contained in the product. It is now common for patients to change from one product to another using a 1:1 lipase ratio and then titrating for maximum efficacy. The North American CF Foundation has published guidelines according to the age of the patient and according to the grams of fat ingested per day [52].
The importance of the correct enzyme ingestion in infants and children is a major concern. There is often difficulty in feeding infants capsules or microspheres however small they may be. The continued use of the unprotected powder enzymes for infants until 1 year of age is common in some centers. In infants, these enzymes may have some advantage over the enteric-coated versions in certain situations. The efficacy of unprotected powder enzymes (in tablet or powder form) has not been directly compared to the enteric-coated version in infants, but infants treated with this approach do achieve growth and weight gain, proving their efficacy [53]. In addition to their use for infants, unprotected powder enzymes are often used to help digest enteral tube feedings where oral administration of enzymes is not possible, or when jejunostomy feeds are required.
Breast-feeding mothers should be instructed on proper infant mouth care after enzyme delivery when the unprotected powder enzymes are provided to these infants. It is recommended that soft cotton swabs or a washcloth dipped in sterile water be used to wipe the inside of the infant’s mouth and inside the gums. This will prove to be sufficient to prevent gum erosion to the infant and nipple irritation to the mother.
For infants with MI requiring surgery or those with an ileostomy, the powder enzymes may provide the advantage of immediate release in the duodenum. Theoretically, this will improve nutrient digestion compared to the pH sensitive, delayed release enteric-coated microsphere enzyme products. Tablets without a protective coating can be crushed if the powder version in capsules or in bottles is not readily available.
The production of specially designed enzymes for the small child has been a recent advancement. A multicenter cross over study in CF infants who were randomized to receive CfC (Creon for children) or regular enzyme for 2-week periods was performed; it compared a spoon administration containing 5000 lipase units with the standard Creon 10,000 capsule [54]. The parental preference was the primary end point; over 75 % of the parents preferred CfC over the standard preparation.
One study demonstrated that there was no improvement when compared to a standard enzyme preparation [55]; another study showed improvement in fat absorption with the bicarbonate-containing enzyme [56]. However, in both studies, approximately 80 % fat absorption was achieved using the bicarbonate-containing enzyme, and Kalnins et al. [55] found the same degree of fat absorption with the conventional enteric-coated enzyme product. Theoretically, the addition of bicarbonate to enteric-coated enzyme preparations might raise the proximal intestinal pH and thereby optimize dissolution of the enteric coating and improve enzyme activity. There were conflicting studies on its efficacy when compared to standard, pH sensitive microsphere enzymes. An enzyme preparation with added bicarbonate had been available in the past, but at time of writing it has not yet received approval by the FDA. Several other enzyme products are in phase 2 or 3 trials. One novel enzyme is liprotamase a non-porcine PERT, containing a biotechnologically derived formulation of crystalline lipase, protease, and amylase [57]. The use of this type of designer drug enzyme has several advantages. The other PERTs are subject to possible viral contamination. In addition, precise dosage standardization had been difficult in the porcine product; the problem of overfill stability has been solved with the new FDA requirements. A preliminary phase 1 study demonstrated good clinical activity, and a multicenter phase 3 study showed that there was significant improvement in the coefficient of fat [56] absorption. A 12-month long-term open-label study [57] of the tolerability and clinical activity in a large number of patients has been published. However, more studies are being carried out on this product. The PERTs currently approved by the FDA have demonstrated efficacy, but new formulations like liprotamase will allow for variety in the type of PERT available.
An important factor for clinicians and families of a CF patient or those with CF to understand and appreciate is that although enzyme therapy for those with PI allows for normal growth and weight gain in most individuals with CF; however, unfortunately, it does not completely correct nutrient malabsorption. There are several reasons for incomplete nutrient digestion with the currently available enzyme products. (1) A proportion of the unprotected powder enzymes or tablets may become inactivated by prolonged exposure to gastric acid. This results in decreased duodenal active enzyme recovery. (2) Enteric-coated microspheres, which dissolve at a pH of > 5.5, may only be released in the ileum if duodenal milieu does not reach this pH as occurs in CF. From prior intubation studies, evidence is confirmed that release of enzymes from enteric-coated microspheres is delayed in CF, and thus, they are delivered beyond the duodenum, even as far distal as the ileum. This results in a nutrient digestion occurring in the more distal small intestine, but not in the duodenum and proximal jejunum as in health. (3) Not only maldigestion but also malabsorption contributes to the insufficient assimilation of nutrients. Fatty acid absorption as well as the digestion of triglycerides is impaired in subjects with CF [58] as suggested by studies done. Other contributing factors to nutrient malabsorption include incomplete lipid solubilization caused by a depleted bile salt pool and thick intestinal mucus. This may affect the unstirred water layer, reducing absorption of fatty acids into the small intestine epithelium. A large center reported < 80 % fat absorption in approximately 30 % of treated patients; therefore, a degree of malabsorption is to be expected for reasons mentioned. Therefore, achieving > 90 % nutrient digestion as evaluated by 72-h fecal fat studies is not likely to occur in a majority of patients with CF.
Patients should not be automatically encouraged to increase their PERT intake if they experience gastrointestinal symptoms such as abdominal pain, bloating, or loose stools, as there are many other etiologies for these symptoms including compliance. For persistent symptoms, a fat balance study should be performed to titrate dose and proton pump inhibitor (PPI) should be considered. After which, if patient does not experience any improvement, investigations for non-pancreatic disease should be explored (see below). Awareness of these factors by both clinicians and patients will help to guide a rational approach to enzyme therapy in CF; an individualized approach to treatment is recommended.
Hepatobiliary Disease
There are a wide variety of hepatobiliary disorders associated with CF (Table 41.2); almost all patients with CF have evidence of hepatobiliary disease. There are no clinical consequences for the vast majority of patients.
Table 41.2
Hepatobiliary complications of CF
Hepatic | Approximate frequency (%) |
|---|---|
Neonatal cholestasis | 5 |
Steatosis | 20–60 |
Focal biliary cirrhosis | 11–70 |
Multilobular cirrhosis | 5–7 |
Liver failure | Rare |
Biliary | |
Microgallbladder | 5–20 |
Distended gallbladder | 3–20 |
Cholelithiasis and sludge | 10–25 |
Intrahepatic sludge/stones | Unknown |
Extrinsic compression of common bile duct | Unknown |
Cholangiocarcinoma | Unknown |
The pathognomonic hepatic feature of CF is focal biliary cirrhosis. Intrahepatic biliary ductal secretion is dependent on CFTR-mediated chloride transport for adequate hydration of the lumen (Fig. 41.2). Loss of CFTR function causes the biliary ductules to become obstructed with thick periodic-acid-Schiff positive material leading to acute and chronic periductal inflammation, bile duct proliferation, and increased fibrosis in scattered portal tracts. Remarkably, adjoining portal tracts are frequently normal. Over 40 years ago, postmortem studies performed show evidence of mild focal disease in 11 % of infants, 27 % of those dying at 1 year, and in more than 70 % of adults. Clinically, significant portal hypertension resulting from severe multilobular cirrhosis develops in only 5–7 % of patients. This is termed “CF-associated liver disease” (CFLD). CFLD is the single most important non-pulmonary cause of death, responsible for 2.5 % of overall mortality in CF. The average age of diagnosis of CFLD is 10–11 years of age, and 90 % are diagnosed before the age of 20 [59] Following routine examination or by laboratory evidence of hypersplenism, most patients are asymptomatic and are often identified by the evidence of hepatosplenomegaly. Hepatocellular function remains well preserved for many years, even decades. Splenomegaly is a consistent finding, and the liver edge is often nodular and hard.
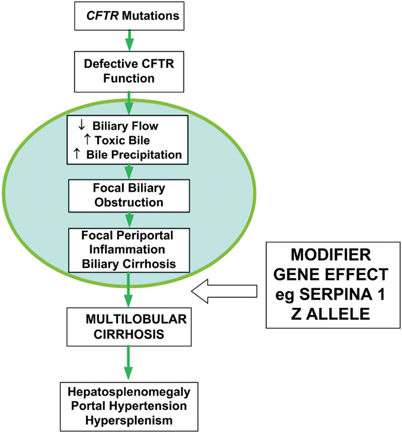
Fig. 41.2
Pathogenesis of cystic fibrosis-related liver disease [32]. Lack of functional CFTR in the biliary ductules causes dehydration, increased concentration, and lack of alkalinization of the duct contents. This causes precipitation of biliary secretions and ductular obstruction. In the majority of patients, the pathologic changes are focal and appear to be clinically inconsequential (circled area). However, approximately 5–10 % of CF patients proceed to develop clinically significant liver disease with evidence of multilobular cirrhosis and portal hypertension . Susceptibility to more severe liver disease may be modulated by other genetic and/or environmental factors. (Reproduced from Ref. [32], Copyright 2007, with permission from BMJ Publishing Group Ltd)
About 40–50 % of CF patients exhibit intermittent elevations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), or gamma glutamyltransferase (GGT), which are generally 1–2.5 times above the upper reference limits. Biochemical markers of liver disease do not reliably identify patients with multilobular cirrhosis nor do they predict the development of end-stage liver disease. Furthermore, patients with advanced multilobular cirrhosis may have normal liver biochemistry test results. As hyperbilirubinemia is rare, elevated serum gammaglobulin level is more likely to be associated with chronic pulmonary inflammation. CFLD occurs predominantly in PI patients. Thus, severe class I, II, or III mutations on both alleles appear to be a risk factor [60]. A male preponderance of severe CFLD has been shown in most studies.
Why a minority of patients with the same severe CFTR mutations progress to CFLD is unclear. However, it has been hypothesized that non-CF modifier genes, such as polymorphisms in genes that upregulate inflammation, fibrosis, or oxidative stress, confer an increased susceptibility. In a multinational gene modifier study of the different candidate genes, the SERPINA 1 Z allele of alpha-1 antitrysin deficiency was found to be strongly associated with CFLD. This result is intriguing as the CF patient requires a double-hit to develop CFLD. Hitherto undefined environmental factors are also likely to be involved.
Gallstones develop in 1–10 % of patients with CF. There is also an increased incidence of variety of intrahepatic and extrahepatic abnormalities of the biliary tree. Nonfunctioning microgallbladders are common, although it is seen less frequently that patients with CF have distended gallbladders appearing obstructed. Stones may also be commonly visualized within the larger intrahepatic and extrahepatic biliary tree. Debray et al. have proposed using the knockout mouse model that the gallbladder itself, rather than the bile duct epithelia may be the major site for a bile acid shunt, the cholecystohepatic shunt, from the gall bladder back to the liver [61].
The principal cause of CF-associated liver disease remaining controversial; it has been postulated that common duct obstruction resulting from extrinsic compression by fibrosis within the head of the pancreas. Following ERCP or transhepatic cholangiographic imaging, changes resembling primary sclerosing cholangitis (i.e., beading and stricturing of the intrahepatic and extrahepatic ducts) are quite commonly observed [62]. These changes are likely due to accumulation of protein, mucus, or sludge within the biliary lumina.
Diagnosis and Management
Studies are being performed to evaluate early liver disease in CF. Clinical examination is necessary to look for signs of chronic liver disease and organomegaly; routine biochemistry is generally not helpful. The evidence of liver disease is often subclinical until CFLD develops as previously stated. As raised serum liver enzymes in a CF patient may be due to other diseases entirely, it is imperative to consider hepatitis (infectious or autoimmune) Wilson disease and other cause of steatosis (malnutrition, diabetes mellitus) as possible causes when reviewing these patients. A small number of infants may present with neonatal cholestasis , in particular those with MI, usually resolving with no long-term affect [63].
In most centers, ultrasonography of the hepatiobiliary system is available, and it includes Doppler measurements of flow in the portal vein; however, the positive predictive value of a normal scan and sensitivity is low. Elastography (Fibroscan R), a new test of liver stiffness (which may be a marker of fibrosis), is used in other chronic liver diseases such as hepatitis C. Early liver disease cannot presently be made on the basis of ultrasound [64].
Recent guidelines by Debray et al. [65] recommend that in order to delay progression of CFLD, ursodeoxychlolic acid treatment should be initiated. In a Swedish study, there was evidence that biochemistry is improved in liver histology [66]; however, this treatment and in particular the recommended dose have been challenged after a trial of high-dose ursodeoxycholic acid in another cholestatic liver disease, Primary sclerosing cholangitis, was terminated due to severe side effects [67]; further studies are obviously needed.
Follow up and management of CFLD are the same as for other chronic liver diseases except for (1) the use of beta blockade prophylaxis for variceal bleeding, it needs to be thoroughly examined in CF patients in consultation with the pulmonologists, and (2) selection criteria for liver transplantation have not been established. Apart from the routine indications, there appears to be evidence that earlier liver transplantation, before the development of significant and irreversible nutritional and pulmonary deterioration, may be helpful. However, large, long-term studies have been difficult to perform [68].
Intestinal Complications
Meconium Ileus
Most patients born with MI present within 24–48 h with evidence of intestinal obstruction with abdominal distension, bilious vomiting, and failure to pass meconium. MI occurs in 15–20 % of CF patients [69] .
Diagnostic aids include a family history of CF and a plain radiograph which may show large distended loops of small bowel and a ground-glass appearance in the right lower quadrant due to due to inspissated meconium in the ileocecal area.
A diagnostic procedure which may be therapeutic is the performance of a contrast enema. The hypertonic enema may lavage the plug of meconium and provided the infant is stable; this procedure may be repeated several times in order to avoid surgery. Surgery is, of course, necessary for complicated MI.
A small proportion presents with perforation in utero with meconium peritonitis and subsequent intra-abdominal calcification which may only be diagnosed incidentally during third trimester ultrasound examinations. Postnatally, up to 50 % of MI patients may have additional problems including malrotation with volvulus or intestinal atresia, termed “complicated MI” .
In a consortium which examined over 3700 cases of MI, several apical transporter genes were found to modify meconium ileu [70], thus appearing that modifier gene(s) together with two severe CFTR alleles confers an increased likelihood of MI. It remains possible that modifier genes with or without environmental factors may explain the variability and severity of other intestinal complications such as distal intestinal obstruction syndrome (DIOS). In a subsequent study, linkage analysis identified a modifier locus for MI on human chromosome 12p13.3 [71].
Distal Intestinal Obstruction Syndrome
Frequently in older children and adults with CF, DIOS a chronic, recurrent form of partial intestinal obstruction occurs. DIOS is frequently confused with other common causes of abdominal pain in patients with CF and almost exclusively in those with PI. Unique to CF, DIOS results from a buildup of adherent, thick intestinal contents in the terminal ileum and proximal colon. The reported frequency is variable, consequently, but a recent study shows a prevalence of 18 % in adults [72]. Intestinal inflammation has been shown in the mouse model [73] and is occasionally shown in humans [74]. Patients usually complain of intermittent episodes of pain, which may or may not be localized to the right lower quadrant. Non-tender, or mildly tender, palpable mass can be felt in the right lower quadrant (although it may not always be localized there). Some patients suffer from intractable chronic pain that is difficult to treat, and on rare occasions, there is complete bowel obstruction.
The European Society for Pediatric Gastroenterology Hepatology and Nutrition (ESPGHAN) Cystic Fibrosis and Pancreatic Disease Working Group defines complete DIOS as the combination of (1) complete intestinal obstruction, as evidenced by bilious vomiting and fluid levels on abdominal radiography with (2) a fecal mass in the ileocecum and (3) abdominal pain and or distension. Incomplete or impending DIOS is defined as (1) a short history of abdominal pain and/or distension (2) a fecal mass in ileocecum but without signs of complete obstruction.
While the exact cause of DIOS is unknown, various precipitating factors have been proposed including abnormal properties of intestinal mucus, dehydration of intraluminal contents, slow intestinal transit, poor compliance with pancreatic enzyme therapy, and a prior history of MI . DIOS should not be confused with simple constipation which is common in CF patients with PI. Although unlike DIOS a right lower quadrant mass is usually not palpable, the rectum will characteristically be full of stool on physical examination, and stooling patterns and consistency are abnormal. Patients undergoing major surgery, such as lung or liver transplant , carry an increased risk of DIOS during the immediate postoperative period.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


