Fig. 36.1
Pathophysiological mechanisms of congenital disorders of intestinal electrolytes transport. 1 Familial diarrhea syndrome is due to a mutation in intestinal guanylate cyclase receptor (GC-C) for the endogenous ligands uroguanylin/guanylin. Ligand binding to GC-C increases intracellular levels of cyclic guanosine monophosphate (cGMP). The cGMP activates cGMP-dependent protein kinase II (PKGII). PKGII phosphorylates the cystic fibrosis transmembrane conductance regulator (CFTR), increasing its Cl–-secreting activity and inhibiting electroneutral NaCl absorption. A missense mutation leads to a gain of function increasing ligand-mediated activation of GC-C and intracellular cGMP levels resulting in chronic secretive diarrhea. 2 Congenital chloride diarrhea is caused by a defect in DRA exchanger leading to severe watery diarrhea due to Cl− malabsorption. The protein has a C-terminal domain, sulfate transporter and antisigma factor antagonist (STAS), that ensures the correct location of the protein on the apical membrane of enterocytes. The STAS domain also interacts with the R-domain of the CFTR, and it is required for CFTR activation. 3 In cystic fibrosis, there is an alternated activity of CFTR. This membrane protein consists of two membrane-spanning domains, two nucleotide-binding domains (NBDs), and a regulatory domain, which controls channel activity. 4 Congenital sodium diarrhea probably derives from a defect in apical membrane Na+/H+ exchangers leading to severe watery diarrhea due to Na+ malabsorption. DRA downregulated in adenoma, DTRL defense terrain research laboratory, cAMP cyclic adenosine monophosphate, AMP adenosine monophosphate, GMP guanosine monophosphate
Two anion exchangers have also been localized to small-intestinal and colonic brush border membranes and cloned: downregulated in adenoma (DRA) and putative anion transporter 1 (PAT1) [1]. DRA was first cloned from colonic mucosa; it was found to be downregulated in villus adenomas and carcinomas and subsequently was found to incur mutations in the rare diarrheal disorder familial chloride diarrhea (see “Congenital chloride diarrhea” below). Both DRA and PAT1 are abundant in the duodenum and present at higher density there than NHE2 and NHE3, suggesting a role in duodenal alkalinization. In the colon, DRA appears to predominate over PAT1.
More than two brush border ion exchangers are required, of course, for the enterocyte to engage in transcellular salt absorption. Increased turnover of the Na +/K +pump and the opening of Cl− and K +channels are also necessary, the latter to counteract associated cell swelling, to permit serosal exit of Cl− taken up from the lumen, and to dissipate the added uptake of K +through the pump.
Diagnostic Approach
The diagnostic approach to CDDs is a multistep process that includes the careful evaluation of the history and clinical data, results of common laboratory, and instrumental procedures and molecular analysis (Fig. 36.2). Positive family history of early onset chronic diarrhea, polyhydramnios, and/or dilated bowel loops at ultrasound examination during pregnancy are highly suggestive of CDDs. The main symptom is chronic diarrhea (lasting longer than 30 days). In the diagnostic approach, it is important to take into account that infections and food allergy are the most common cause of diarrhea also at this particular age [4], and that these conditions together with malformations of gastrointestinal tract, should be considered as primary diagnostic hypotheses in a infant with chronic diarrhea [4]. However, in all cases of chronic diarrhea starting in the first weeks of life, the presence of a congenital disorder of intestinal electrolyte transport should be excluded. A fundamental step in the diagnostic process is the identification of an osmotic or secretory mechanism leading to diarrhea. In osmotic diarrhea, unabsorbed luminal substances are responsible for accumulation of fluids in intestinal lumen and diarrhea significantly improves during fasting, whereas in secretory diarrhea, fluids are actively secreted in the intestinal lumen and diarrhea continues during fasting [5]. Furthermore, the determination of stool electrolyte concentration and fecal ion gap is important to discriminate the two mechanisms responsible for CDDs (Fig. 36.1). If ion gap is > 50 fecal, osmolarity derived from ingested osmotically active nutrient or not measured ion (i.e., Cl−, Mg2+…). In contrast, a low osmotic gap (< 50) is typically observed in secretory diarrhea. A direct measurement of Na+ and Cl− concentration in the stool may clearly suggest a diagnosis of congenital sodium diarrhea (CSD) or chloride losing diarrhea (CLD), respectively [5]. When an osmotic mechanism is suspected, the next step of laboratory investigation includes blood gas, blood glucose, ammonium, albumin, triglycerides and cholesterol, aminoaciduria and the search of reducing substances in the stools, steatocrit, and sweat test (Fig. 36.2). CF is suggested by the presence of steatorrhea and confirmed by a positive sweat test. On the other hand, signs of inflammation in children with secretory diarrhea and familiar history of chronic diarrhea justify the suspect of a familial diarrhea syndrome (FDS). Recent availability of molecular analysis for many of these conditions has progressively limited the need for invasive procedures (Fig. 36.2).
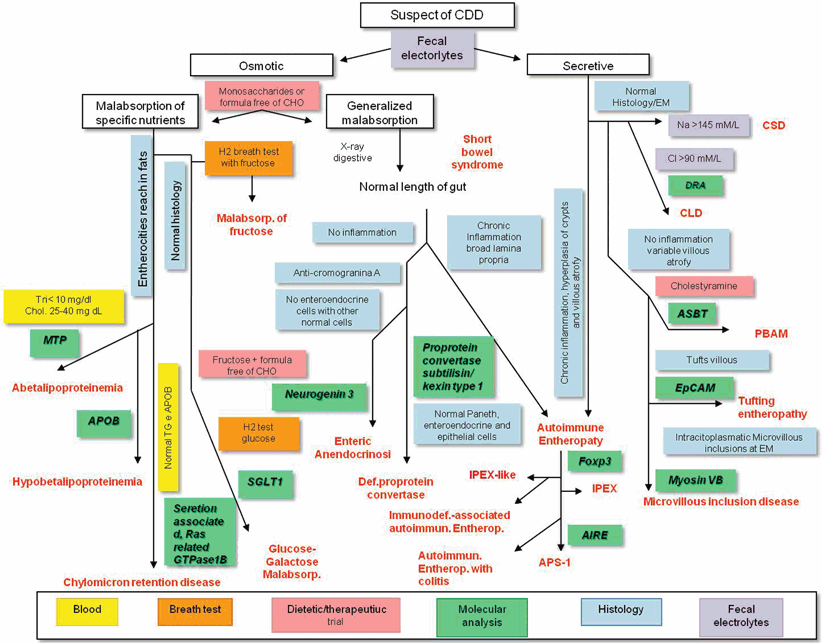
Fig. 36.2
Indications for an integrated diagnostic approach to the main forms of CDDs. The diagnostic approach is a multistep process that includes the evaluation of anamnesis and clinical data, results of common laboratory and instrumental procedures and molecular analysis . The fundamental step in the diagnostic process of CDDs is the identification of an osmotic or secretory mechanism leading to diarrhea. Moreover, the determination of stool electrolyte concentration and fecal ion gap is important to discriminate the two mechanisms responsible for CDDs. The next step is laboratory investigation includes blood gas, blood glucose, ammonium, albumin, triglycerides, and cholesterol, aminoaciduria and the search of reducing substances in the stools, steatocrit, and sweat test. Finally, intestinal biopsy with histologic examination is crucial for the diagnosis of most CDDs. Molecular analysis, when available, is important and could limit invasive procedures CDD congenital diarrheal disorder, DRA downregulated in adenoma, CLD congenital chloride diarrhea, ASBT apical sodium-dependent bile acid transporter, EpCAM epithelial cell adhesion molecule, PBAM peripheral blood adherent monocyte, IPEX immune dysregulation, polyendocrinopathy, enteropathy, X-linked, MTP media transfer protocol, APOB apolipoprotein B, SGL2 sodium-glucose transport, AIRE autoimmune regulator
Congenital Chloride Diarrhea
CLD (OMIM 214700) is caused by a mutation in the solute carrier family 26 member 3 (SLC26A3) gene, and it is responsible for a life-long watery diarrhea with high fecal Cl− concentration [6]. The gene, located on chromosome 7.q31, encodes for a transmembrane protein that takes part in gut Cl−/NaHCO3 − exchange (Fig. 36.2) [7]. The SLC26A3 is located close to the gene encoding for the cystic fibrosis transmembrane conductance regulator (CFTR , raising a possibility of CFTR modulation in the pathogenesis of CLD [8]. The protein encoded by SLC26A3 gene has a C-terminal domain, STAS (sulfate transporter and antisigma factor antagonist), that ensures the correct location of the protein on the apical membrane of enterocytes. Mutations determine transporter mistrafficking and cytosol retention [9]. The STAS domain also interacts with the R-domain of the CFTR, and it is required for CFTR activation. Studies have shown that deglycosylation of HA-SLC26A3 may contribute to the pathogenesis of diarrhea associated with congenital disorder of glycosylation (CDG) because oligosaccharides protect HA-SLC26A3 from proteolytic digestion enzymes in the intestine [10]. The gene SLC26A3 has also extraintestinal expression: it is expressed in sweat glands, the male reproductive tract, and kidney, while it is downregulated in human colon adenomas and adenocarcinomas [11–13] .
The majority of the SLC26A3 mutations are single-nucleotide substitutions (nonsense, frameshift, and missense), while additional mutations as deletions/insertions are minor. There is no evidence of a correlation between genotype and phenotype. It has been demonstrated that an identical genetic background of CLD may show different clinical course [14] or different responses to therapy [15].
Countries with a highest incidence are Finland (1:35,000), Poland (1:200,000), Kuwait, and Saudi Arabia [16]. In Arabian countries, the high incidence is mostly due to consanguineous marriages [16]. There is a single mutation in the above-mentioned ethnic groups: in Finns, the p.V317del mutation affects up to 90 % of CLD alleles; in Saudi Arabians and Kuwaitis, pG187X is present in more than 90 % of altered chromosomes; in Poles, 50 % of CLD alleles carry the p.1675dup [6].
The main clinical symptom is lifelong watery diarrhea with high Cl− content and low pH [1]. If not treated, diarrhea leads to dehydration, metabolic alkalosis, hyponatremia, hypokalemia, hypochloremia, and failure to thrive. Because of the intrauterine onset of diarrhea, CLD pregnancies are characterized by polyhydramnios, and the newborn could be mild premature [16, 17]. Postnatally, CLD patients present prominent abdominal distention and dehydration [16, 18].
Suspect of CLD in pregnant women arises with maternal polyhydramnios, lack of meconium, and images of distended loops of ileum. These aspects, evidenced by ultrasonography, are an indication of intrauterine onset of diarrhea. The abdomen is usually large and distended, and intestinal contractions are visible. Loops of ileum and colon are dilated with air and fluid, and ascites is often present. Hyperbilirubinemia is common and is caused in part by the dehydration. It is worth remembering that excessive volume and salt depletion reduce the amount of diarrhea and may result in a low fecal Cl− of even 40 mmol/L such as a stool dilution by urine [16]. After correction of electrolyte homeostasis by salt substitution, the next thing to do is measuring Cl− on a stool sample, possibly obtained with a soft catheter to avoid urine contamination. A fecal Cl− > 90 mmol/L confirms the diagnosis in most cases and leads to molecular analysis of SLC26A3. Untreated patients show retarded growth and development, with mental and psychomotor delay .
The main therapeutic approach is based on substitutive therapy with NaCl/KCl (see Table 36.1) [19]. Therapy with the short-chain fatty acid butyrate is also beneficial in patients affected by CLD [20, 21]. Butyrate could reduce mistrafficking or misfolding of DRA and may enhance gene expression through the activation of a region crucial for high-level transcription. Butyrate is also able to modulate transepithelial ion transport through the stimulation of Na+/H+ exchangers 2 (NHE2) and 3 (NHE3) activity and inhibition of Cl− secretion by limiting the action of the cotransporter Na–K–2Cl (encoded by NKCC1) on enterocyte basolateral membrane. In fact, treatment with oral butyrate (100 mg/kg/day) allows a progressive reduction to normal in the number of bowel movements and stool volume, an improvement in stool consistency, and a reduction of fecal incontinence episodes [15]. The effect of butyrate on stool pattern is evident within the first 48 h and remains stable during the following days of treatment. Depending on genotype, the response to butyrate may vary, and this explains the different benefits on the basis of ethnicity [15]. Because missense and deletion mutations allow the expression of SLC26A3 at membrane level, a full response to butyrate (defined by a concomitant significant reduction of Na + and Cl− fecal losses and improvement in stool pattern) is observed only in patients with these types of mutation. On the contrary, a partial response is to be expected in patients with nonsense or splicing mutations [15] .
Table 36.1
Therapeutic approach to a child with congenital chloride diarrhea
Treatment of acute dehydration | |||
|---|---|---|---|
1. Initial treatment of fluid depletion (over 6–8 h) IV infusion of 0.9 % NaCl solution, NO fluids containing bicarbonate 120–500 mL/day for children under 7 years 500–1000 mL/day for children over 7 years | |||
2. Maintenance therapy (over 24 h) Intravenous 5 % glucose 100 mL pro kg (children weight 10 kg) + 50 mL pro kg for each additional kg (children weight between 11 and 20 kg) + 20 mL pro kg for each additional kg (children weight 20 kg) For adult patients: 2000–2500 mL/day NaCl 20 mmol/L added KCl maintenance need + calculated K + depletion added (50 mmol/L) If severe hypokalemia, higher KCl doses of even 70 (−100) mmol/L | |||
3. Replacement of ongoing losses for diarrhea (0.9 % NaCl and, if necessary, KCl) | |||
Salt substitutive therapy in stable clinical condition | |||
Small children (0–3 years) | Older children | Adolescents and adults | |
NaCl | 0.7 % (7 g/L; 120 mmol/L)a | 1.8 % (18 g/L; 308 mmol/L) | Equal molar ratios of NaCl and KCl |
KCl | 0.3 % (3 g/L; 40 mmol/L)a | 1.9 % (19 g/L; 255 mmol/L) | Equal molar ratios of NaCl and KCl |
Concentration of Cl− | 160 mmol/L | 563 mmol/L | – |
Need for Cl− | 6–8 mmol/kg/day | 3–4 mmol/kg/day | 3–4 mmol/kg/day |
Administration | Intravenousb/Oral | Oral | Oral |
Salt dosage | Ready-made solution | Ready-made solution | Dose bags |
Doses per day | 3–4 | 2–3 | 2–3 |
Pharmacological treatment | |||
Oral sodium butyrate, 100 mg/kg/day, divided in two doses (particularly useful in patients with missense or deletion mutations) | |||
Long-term prognosis of CLD patients is generally favorable. All of the patients treated adequately reach adult life because oral salt substitution with NaCl and KCl allows normal growth and development [17, 19]. Complications such as intestinal inflammation , renal disease, hyperuricemia, inguinal hernias, spermatoceles, and male subfertility are possible [17, 19]. Untreated or poorly treated disease is associated with impaired renal function and nephrocalcinosis, and even with end-stage renal disease [12].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


