Primary CIPO
Neuropathy post neonatal necrotizing enterocolitis [57]
Secondary CIPO
Conditions affecting GI smooth muscle
Other (celiac disease, eosinophilic gastroenteritis, Crohn’s disease , radiation injury, Chagas disease, Kawasaki disease, angioedema, mitochondrial disorders, drugs, e.g., opiates, anthraquinone laxatives, calcium channel blockers, antidepressants, antineoplastic agents, e.g., vinca alkaloids, paraneoplastic CIPO, major trauma/surgery, chromosome abnormalities) [107–133]
Idiopathic
Etiology and Pathophysiology
The integrity of GI sensorimotor function relies on precise coordination between the autonomic nervous system, enteric nervous system (ENS), ICC, and smooth muscle cells. Any noxious stimulus, irrespective of its origin and etiology, that affects the neuromuscular elements and control of GI tract can lead to impaired peristalsis and the stasis of luminal contents [1]. A variety of disorders and pathophysiological mechanisms can potentially affect the structure or function of the neuromuscular elements of the GI tract and lead to CIPO (Table 23.1) [1]. Neurological (e.g., multiple endocrine neoplasia (MEN) type IIb, familial dysautonomia) and metabolic (e.g., diabetes mellitus) conditions may affect the extrinsic GI nerve supply [19]. Neurotropic viruses may evoke an inflammatory process targeting both the ENS and extrinsic neural pathways [93]. Paraneoplastic syndromes may also exert a destructive effect on the ENS by initiating an inflammatory process that targets the neurons of ganglia located in the submucosal and myenteric plexuses. This is mediated by both a cellular infiltrate and production of circulating antineuronal antibodies [19, 134]. Some pathologies (e.g., muscular dystrophy) may target enteric smooth muscle fibers, whereas others such as dermatomyositis, scleroderma, Ehlers–Danlos syndrome, radiation enteritis may distort both ENS and gut smooth muscle leading to a mixed neuromyopathic disorder [12, 135, 136]. Finally, although entities such as celiac disease, hypothyroidism, hypoparathyroidism, and pheochromocytoma presumably cause CIPO by affecting the GI neuromuscular integrity, the exact mechanism is not fully understood.
Genetics
Elucidation of the genetic basis of CIPO has been somewhat disappointing. Some familial cases of CIPO have been recognized but there appear to be several patterns of inheritance, perhaps reflective of the great heterogeneity of CIPO conditions. Both autosomal dominant and recessive modes of inheritance have been described for neuropathic and myopathic types of CIPO [5, 13, 14, 135, 137]. More specifically, rare autosomal dominant mutations in the SOX10 gene, which encodes a transcription factor important in ENS development, result in a CIPO clinical phenotype along with features such as sensorineural deafness and pigmentary anomalies [138, 139]. Homozygosity on the region 8q23–q24 has been implicated in the pathogenesis of an autosomal recessive form of CIPO characterized by severe GI dysmotility, Barrett’s esophagus, and cardiac anomalies [140, 141].
X-linked inheritance (locus Xq28) with recessive transmission has been described in CIPO [15, 142, 143]. Mutations of filamin A (FLNA) and L1 cell adhesion molecule (L1CAM) genes, which are both located on chromosome Xq28, result in predominantly myopathic and neuropathic forms of CIPO, respectively. Additional involvement of the central nervous system, heart (patent ductus arteriosus), and blood (thrombocytopenia) in both conditions has also been described [143–145].
Mutations in mitochondria are increasingly implicated in CIPO. Mutations in the thymidine phosphorylase gene ( TYMP, also termed as endothelial cell growth factor-1, ECGF1), or in the polymerase-γ gene (POLG) result in recessive myopathic forms of CIPO. The former is the cause of MNGIE, whereas the latter leads to a form without encephalopathy. Apart from the GI dysmotility, MNGIE is characterized by severe malnutrition, opthalmoplegia, and leucoencepalopathy on brain MRI [146–148].
The responsible genes for familial visceral neuropathy and myopathy are yet to be identified.
Histopathology
In adults, GI histology is reported to be normal in approximately 10 % of CIPO cases, while in the experience of the authors, this figure is likely to be higher in children. However, its role in CIPO remains crucial and therefore an adequate full-thickness bowel biopsy (preferably a circumferential sleeve of at least 1–2 cm) is recommended whenever surgery is being considered [8, 26, 149]. Recent initiatives are addressing a more standardized and hopefully effective histological approach to diagnosis in GI motility disorders such as CIPO [Knowles et al., working group] [25, 150, 151].
On the basis of histology , CIPO is classified into neuropathy, myopathy, or mesenchymopathy [25]. However, mixed forms (e.g., neuromyopathy) are also recognized [25, 152, 153].
Neuropathies and myopathies can be further subdivided into inflammatory and degenerative. Inflammatory neuropathies are characterized by an infiltration of T lymphocytes and plasma cells in the myenteric plexuses (myenteric ganglionitis) and neuronal axons (axonopathy) [25, 154]. It has been proposed that five or more lymphocytes per ganglion are required for the diagnosis of myenteric ganglionitis [25]. Of note, patients with lymphocytic infiltration of the myenteric plexuses may also develop increased titers of antinuclear antibodies (ANNA-1/anti-Hu, anti-voltage-gated potassium channel or VGKC) [155–157]. These immunologic responses may result in neuronal degeneration and loss by activating apoptotic and autophagic mechanisms [156, 158]. Infiltration of the myenteric ganglia with other cells such as eosinophils and mast cells has been described but their exact clinicopathological significance is yet to be clarified given limited data [159–162]. All these data support the role of the immune system in the pathogenesis of inflammatory CIPO [163].
Degenerative neuropathies are poorly understood given the limited amount of available data [164–166]. Main histopathologic characteristics of this group include a decrease in the number of intramural neurons along with changes in nerve cell bodies and axons [150, 154]. It has been postulated that apoptotic mechanisms are involved in the degenerative process potentially caused by aberrant calcium signaling, mitochondrial disorders, production of free radicals, and abnormalities in the function of glial cells [150, 152, 167, 168].
Similarly to neuropathies, myopathies are also divided into inflammatory and degenerative. Inflammatory myopathies, also reported by the term leiomyositis, are characterized by infiltration of T lymphocytes into both the circular and longitudinal enteric muscle layers. This process if not treated appropriately with immunosuppressive agents may lead to a severe clinical picture of CIPO [44, 169].
The histopathologic findings in degenerative myopathies include smooth muscle fiber vacuolization and fibrosis [170, 171]. Diverticula may also be present especially if the longitudinal muscle coat is more affected compared to the circular muscle layer [146, 148].
Novel immunohistochemical techniques such as smooth muscle markers, namely, smoothelin, smooth muscle myosin heavy chain, and histone deacetylase 8, may reveal histiopathologic subtleties otherwise not detectable with conventional immunostaining and histochemistry methods [172].
Mesenchymopathies are defined by ICC abnormalities (decreased density of ICC network, intracellular abnormalities) and have been demonstrated in CIPO patients [150, 173]. Although sufficient data exist regarding their role in the pathogenesis of diabetic gastroparesis, further research is required regarding ICC involvement in the etiopathogenesis of other GI motility disorders [25].
Clinical Picture
In a few cases, the diagnosis of CIPO is suggested in utero by ultrasonographic findings of polyhydramnios, abdominal distention, and megacystis; however, the majority of cases present in the neonatal period or early infancy [8, 26]. The symptomatology varies according to the age at diagnosis and the part of the GI tract, which is primarily affected. Approximately, one-third of children with congenital CIPO (myopathic and neuropathic) have intestinal malrotation [22]. Cardinal signs and symptoms of CIPO include those of obstruction, namely abdominal distention (88 %), vomiting (72 %, which can be bilious), and constipation (61 %). Abdominal pain (44 %), failure to thrive (31 %), and diarrhea (28 %) may also be part of the clinical picture [8, 9, 149].
Dehydration (which can be severe) and malnutrition are often underdiagnosed especially given that weight can be an unreliable measure due to pooling of significant volumes of fluid (third spacing) within distended gut loops. Intraluminal gut content stasis can also lead to small bowel bacterial overgrowth which can further exacerbate symptoms of diarrhea and abdominal distention [149].
CIPO may also manifest with extraintestinal signs and symptoms, such as recurrent urinary tract infections or neurologic abnormalities [17, 147]. Furthermore, patients may complain of symptoms indicative of an underlying disorder that accounts for secondary CIPO (e.g., proximal muscle weakness in dermatomyositis) [59].
The clinical course of CIPO is characterized by exacerbations and remissions, where the former can be precipitated by a number of factors such as surgery, general anesthesia, infections, and emotional stress [26]. In the most severe cases, the natural course of the disease leads to worsening intestinal function and ultimately to intestinal failure [9, 149]. This is especially true in cases where the diagnosis and/or institution of appropriate treatment has been delayed.
Diagnosis
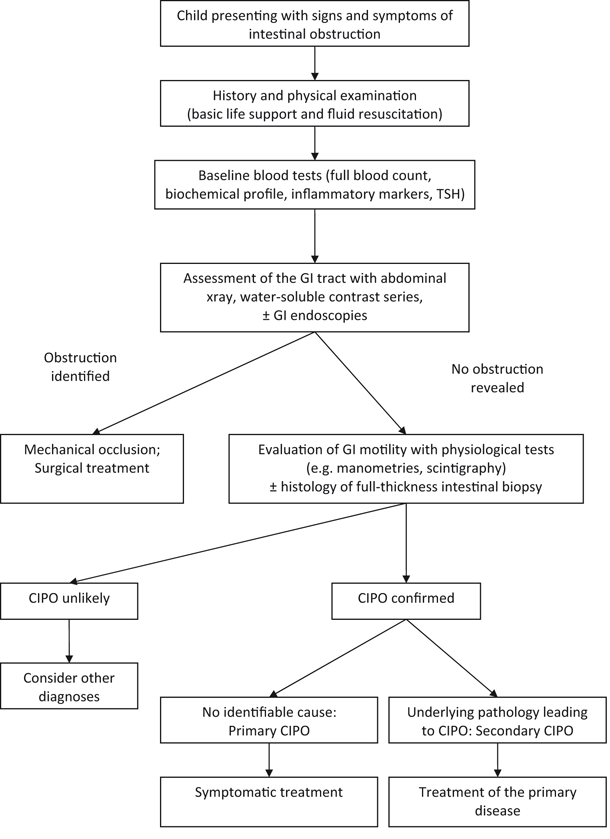
CIPO should be suspected in children with early onset, chronic, recurrent, or continuous signs of intestinal obstruction especially where a surgical cause cannot be established (e.g., repeated “normal” exploratory laparotomies). The diagnosis of CIPO should follow a structured algorithm. Although a detailed history, clinical examination, and laboratory tests may suggest the presence of CIPO, or help elucidate its cause, the definitive diagnosis should rely on the use of targeted investigations to (i) exclude mechanical occlusion of the gut lumen, (ii) confirm GI dysmotility, and (iii) rule out treatable causes.
Careful clinical history and physical examination may help in defining the onset, the severity and progression of the disease, and the part of the GI tract primarily affected, and they also provide useful information regarding associations (e.g., family history), potential secondary causes (e.g., medications), and complications (e.g., dehydration). Laboratory tests (e.g., serum electrolytes, thyroid-stimulating hormone (TSH), lactic acid, specific autoantibodies) are useful in cases of secondary CIPO and assessing the clinical state of the patients admitted acutely or undergoing a diagnostic protocol.
The definitive diagnosis of CIPO in children is often reliant on a number of diagnostic tests, which exclude luminal obstruction and confirm the presence of impaired GI motility. These are discussed below .
Imaging
Plain abdominal radiographs may demonstrate a dilated GI tract, with air-fluid levels, whereas contrast GI series can reveal anatomical abnormalities (e.g., malrotation, microcolon) and exclude the presence of gut occlusive lesions (Fig. 23.1a) [2, 149, 174]. It needs to be kept in mind that a water-soluble substance should be used instead of barium in order to prevent flocculation and inspissation of the contrast material.
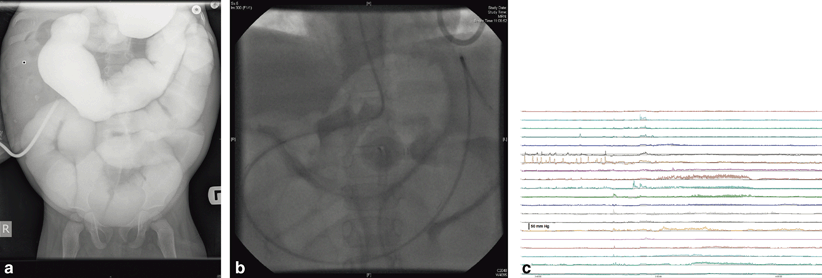
Fig. 23.1
Investigation findings of a 3-year-old boy with a history of recurrent episodes of abdominal distension and vomiting since the neonatal period, and now showing a marked reduction in enteral feed tolerance. a Contrast follow-through study (administered via gastrostomy) showing filling of grossly dilated small intestinal loops, without any apparent hold up or change in calibre. b Plain abdominal radiograph taken following placement of antroduodenal manometry catheter into the same patient performed under fluoroscopic guidance. The tip of the catheter has been advanced beyond the duodenojejunal junction to facilitate optimal manometric recording of both the stomach and small intestine. c Antroduodenal manometry tracing from patient showing the presence of some gastric antral contractions and a migrating motor complex (phase III activity) passing down the small intestine. The amplitude of small intestinal contractile activity is very low (not exceeding 20 mmHg) suggesting a diagnosis of myopathic chronic intestinal pseudo-obstruction
Novel imaging modalities such as cine MRI have been recently performed with promising results in adult series but there are no data regarding their applicability and usefulness in pediatrics [175].
Endoscopy
Endoscopy may identify fore- or hindgut mechanical occlusion previously missed on radiology, and it allows duodenal biopsies to exclude mucosal inflammation [176]. Novel techniques (e.g., natural orifice transluminal endoscopic surgery—NOTES) may revolutionize the role of endoscopy in the diagnosis of gut motility disorders by providing the ability of full-thickness biopsy sampling in a safe and minimally invasive way [177, 178].
Motility Investigations
These studies are performed to assess the GI motility and to define the underlying pathophysiologic process, and these studies form the hallmark of diagnosis in pediatrics. Investigations include GI manometries (esophageal, antroduodenal, colonic, anorectal), GI scintigraphy (e.g., gastric emptying, colonic transit), electrogastrography, and radiopaque markers. The usefulness of novel technologies, such as SmartPill, remains to be determined [8, 179, 180].
Although in children with CIPO the involvement of GI may be generalized, the small intestine is always affected; thus, antroduodenal manometry remains the most discerning test, and its optimal placement is pivotal (Fig. 23.1b) [181]. Neuropathic cases manifest with uncoordinated contractions, which are of normal amplitude, whereas in myopathic CIPO, motor patterns have normal coordination; however, the amplitude of intestinal contractions is low (Fig. 23.1c) [182–184]. Additionally, manometry may facilitate the dynamic assessment of potential pharmacotherapeutic options and feeding strategies (e.g., feasibility of oral or enteral feeds) as well as indicate disease prognosis [185–187].
In the most challenging cases, exploratory surgery (laparotomy or laparoscopic-assisted procedures) may be required to definitively exclude mechanical obstruction from CIPO. One however should bear in mind that surgery may precipitate a pseudo-obstructive episode and may also lead to adhesions formation, which can further complicate future diagnostic or therapeutic procedures. Where possible, investigations and then diagnostic/therapeutic surgery should be performed in timeline sequence and in referral center.
Histopathology along with genetics can also be very useful in establishing or confirming the diagnosis of CIPO, highlighting the underlying pathophysiologic process, thus aiding the overall management.
Differential Diagnosis
CIPO has to be differentiated from mechanical obstruction; the latter is usually characterized by marked abdominal pain (in keeping with the abdominal distention), specific radiologic signs, and manometric patterns [188, 189]. Acute functional obstruction (e.g., postoperative ileus), functional GI disorders (e.g., rumination syndrome), and pediatric condition falsification should be considered and appropriately investigated and managed [149, 190, 191].
Treatment
The therapeutic approach in CIPO is threefold as it aims to (i) preserve growth and development by maintaining adequate caloric intake, (ii) promote GI motility with combined medical and surgical interventions, and (iii) treat disease-related complications or underlying pathologies that cause secondary CIPO. Despite the limited effects of the currently applied therapeutic modalities, refinements and evolution in nutritional, medical, and surgical strategies have considerably improved the overall management of CIPO [136, 192]. Acute management of episodes of pseudo-obstruction are generally treated conservatively by nil by mouth, intravenous fluid, and drainage of stasis through nasogastric (NG) tube or preformed ostomies. Careful attention to fluid and electrolytes is imperative.
Nutrition
The role of nutrition in CIPO is of paramount significance as it is well known that gut motility improves with optimal nutritional support and declines in the face of under- or malnutrition [8]. In the long term, approximately one-third of pediatric CIPO patients require either partial or total parenteral nutrition, another third require a degree of intragastric or intra-enteral feeding, whereas the remaining children are able to tolerate sufficient oral nutrition. However, within all of groups, patients able to tolerate feeds may require some dietary modification in order to maintain enteral nutrition and avoid bezoar formation (e.g., bite and dissolvable feeds, restriction diets, hydrolyzed formula). Although parenteral nutrition is lifesaving, it is associated with significant risk of complications, such as central line infections and liver disease, and therefore maintaining patients on maximally tolerated enteral nutrition is always strongly encouraged [26]. In the more severe CIPO cases, continuous rather than bolus feeds administered via a gastrostomy or jejunostomy may be better tolerated particularly in children with impaired gastric motor function [193–195].
Medications
The therapeutic role of drugs in CIPO patients is mainly limited to the control of intestinal inflammation, suppression of bacterial overgrowth, and promotion of GI motility [186, 195].
Prokinetics (e.g., metoclopramide, domperidone, erythromycin, azithromycin, octreotide, neostigmine) usually combined with antiemetics (e.g., promethazine, ondansetron) have been used in an attempt to improve the GI motor function and reduce the severity of nausea and vomiting [196–199]. The use of some of these agents is limited by variable efficacy and unacceptable extraintestinal side effects (e.g., metoclopramide, neostigmine). The best-studied and tested prokinetics, that is, cisapride and tegaserod have been withdrawn from the market due to safety concerns [200]. The need for new prokinetics with increased safety and efficacy has resulted in new products (e.g., prucalopride, aprepitant, ghrelin), but there are limited data of their use in pediatric CIPO, further impacted on by restricted availability and licensing [201–203]. Undoubtedly, current medical regimens for CIPO are based on limited literature and/or expert opinion (e.g., combined use of octreotide and erythromycin) and are yet to be tested in future in the context of controlled trials [186, 204].
Surgery
Surgery remains a valuable intervention on patients with CIPO as it has a multidimensional role in both the diagnostic (e.g., full thickness biopsies) and therapeutic processes (e.g., insertion of feeding tubes, formation of decompressing ostomies such as gastrostomy, ileostomy) [195, 205, 206].
Indeed, adequate bowel decompression is crucial not only in providing symptomatic relief by reducing the frequency and the severity of pseudo-obstructive episodes but also in limiting further deterioration of the intestinal motor activity secondary to chronic distention, and in enhancing the tolerance of enteral feeding [195, 205]. Long decompression enteral tubes and extensive bowel resections are approaches mainly reported in adult CIPO cohorts but remain untested in terms of practicality, efficacy, and safety in pediatrics [207, 208]. Rate of significant surgical complications, such as stoma prolapse, infection, and leakage can be significant.
Novel surgical methods involve implantation of devices providing electrical pacing of the GI neuromusculature, but data in children are scanty and limited [209].
Small bowel transplantation remains the only definitive cure. Recent advances in both surgical techniques (e.g., multivisceral transplantation) and immunosuppression strategies have resulted in improved outcomes and survival as reported by centers with the relevant expertise showing a survival rate of 50 % at 3 years [210–213].
Natural History and Prognosis
Both pediatric and adult CIPO have a severe clinical course, characterized by repetitive relapses and remissions. Unfortunately, the low index of suspicion among physicians along with lack of well-defined diagnostic criteria and readily available facilities in performing specialized diagnostic tests (e.g., manometry) often accounts for delays in the diagnosis and repetitive unnecessary investigations and surgery [13–15, 164].
The majority of the patients complain of symptoms, which progressively worsen and impact upon the tolerance of enteral nutrition and increasing reliance on total parenteral nutrition. The latter in conjunction with disease-related adverse events (e.g., central line infections, impairment of the liver function, immunosuppression after small bowel transplantation, surgical procedures) accounts for high morbidity, poor quality of life, and mortality rates up to 30 % [11, 21, 28, 214–218].
Despite recent diagnostic and therapeutic advances, CIPO in children remains a serious, life-threatening disease with significant impact on the well-being not only of patients themselves but also of their families [218].
Summary
Childhood CIPO is an enigmatic disease with poorly defined etiopathogenesis, which is reflected on the limitations encountered in both the diagnostic process and therapeutic management. Clearly, multinational initiatives are required to raise awareness, establish stringent diagnostic criteria, and evolve current therapeutic modalities.
Acknowledgments
Nikhil Thapar is supported by Great Ormond Street Hospital Children’s Charity.
References
1.
Gabbard SL, Lacy BE. Chronic intestinal pseudo-obstruction. Nutr Clin Pract. 2013;28:307–16.PubMed
2.
Rudolph CD, Hyman PE, Altschuler SM, et al. Diagnosis and treatment of chronic intestinal pseudo-obstruction in children: report of consensus workshop. J Pediatr Gastroenterol Nutr. 1997;24:102–12.PubMed
3.
Dudley HA, Sinclair IS, Mc LI, et al. Intestinal pseudo-obstruction. J R Coll Surg Edinb. 1958;3:206–17.PubMed
4.
Naish JM, Capper WM, Brown NJ. Intestinal pseudoobstruction with steatorrhoea. Gut. 1960;1:62–6.PubMedCentralPubMed
5.
Stephens FO. Syndrome of intestinal pseudo-obstruction. Br Med J. 1962;1:1248–50.PubMedCentralPubMed
6.
Byrne WJ, Cipel L, Euler AR, et al. Chronic idiopathic intestinal pseudo-obstruction syndrome in children—clinical characteristics and prognosis. J Pediatr. 1977;90:585–9.PubMed
7.
Schuffler MD, Pope CE 2nd. Studies of idiopathic intestinal pseudoobstruction. II. Hereditary hollow visceral myopathy: family studies. Gastroenterology. 1977;73:339–44.PubMed
8.
Hyman P, Thapar N. Gastrointestinal motility and functional disorders in children. In: Faure C, Di Lorenzo D, Thapar N, editors. Pediatric neurogastroenterology. New York: Springer; 2013. pp. 257–70.
9.
Thapar N. Clinical picture of intestinal pseudo-obstruction syndrome. J Pediatr Gastroenterol Nutr. 2011;53(Suppl 2):S58–9.PubMed
10.
Amiot A, Joly F, Cazals-Hatem D, et al. Prognostic yield of esophageal manometry in chronic intestinal pseudo-obstruction: a retrospective cohort of 116 adult patients. Neurogastroenterol Motil. 2012;24:1008–e542.
11.
Vargas JH, Sachs P, Ament ME. Chronic intestinal pseudo-obstruction syndrome in pediatrics. Results of a national survey by members of the North American Society of Pediatric Gastroenterology and Nutrition. J Pediatr Gastroenterol Nutr. 1988;7:323–32.PubMed
13.
Stanghellini V, Cogliandro RF, De Giorgio R, et al. Natural history of chronic idiopathic intestinal pseudo-obstruction in adults: a single center study. Clin Gastroenterol Hepatol. 2005;3:449–58.PubMed
14.
Amiot A, Joly F, Alves A, et al. Long-term outcome of chronic intestinal pseudo-obstruction adult patients requiring home parenteral nutrition. Am J Gastroenterol. 2009;104:1262–70.PubMed
15.
Lindberg G, Iwarzon M, Tornblom H. Clinical features and long-term survival in chronic intestinal pseudo-obstruction and enteric dysmotility. Scand J Gastroenterol. 2009;44:692–9.PubMed
16.
Iida H, Ohkubo H, Inamori M, et al. Epidemiology and clinical experience of chronic intestinal pseudo-obstruction in Japan: a nationwide epidemiologic survey. J Epidemiol. 2013;23:288–94.PubMedCentralPubMed
17.
Mc Laughlin D, Puri P. Familial megacystis microcolon intestinal hypoperistalsis syndrome: a systematic review. Pediatr Surg Int. 2013;29:947–51.PubMed
18.
Blondon H, Polivka M, Joly F, et al. Digestive smooth muscle mitochondrial myopathy in patients with mitochondrial-neuro-gastro-intestinal encephalomyopathy (MNGIE). Gastroenterol Clin Biol. 2005;29:773–8.PubMed
19.
De Giorgio R, Cogliandro RF, Barbara G, et al. Chronic intestinal pseudo-obstruction: clinical features, diagnosis, and therapy. Gastroenterol Clin North Am. 2011;40:787–807.PubMed
20.
Mousa H, Hyman PE, Cocjin J, et al. Long-term outcome of congenital intestinal pseudoobstruction. Dig Dis Sci. 2002;47:2298–305.PubMed
21.
Heneyke S, Smith VV, Spitz L, et al. Chronic intestinal pseudo-obstruction: treatment and long term follow up of 44 patients. Arch Dis Child. 1999;81:21–7.PubMedCentralPubMed
22.
Streutker CJ, Huizinga JD, Campbell F, et al. Loss of CD117 (c-kit)- and CD34-positive ICC and associated CD34-positive fibroblasts defines a subpopulation of chronic intestinal pseudo-obstruction. Am J Surg Pathol. 2003;27:228–35.PubMed
23.
Jain D, Moussa K, Tandon M, et al. Role of interstitial cells of Cajal in motility disorders of the bowel. Am J Gastroenterol. 2003;98:618–24.PubMed
24.
Struijs MC, Diamond IR, Pencharz PB, et al. Absence of the interstitial cells of Cajal in a child with chronic pseudoobstruction. J Pediatr Surg. 2008;43:e25–9.PubMed
25.
Knowles CH, De Giorgio R, Kapur RP, et al. The London Classification of gastrointestinal neuromuscular pathology: report on behalf of the Gastro. 2009 International Working Group. Gut. 2010;59:882–7.PubMed
26.
Hyman P. Chronic intestinal pseudo-obstruction. In: Wyllie R, Hyams J, Kay M, editors. Pediatric gastrointestinal and liver disease. Philadelphia: Elsevier; 2011. pp. 505–11.
27.
Puri P, Shinkai M. Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg. 2005;14:58–63.PubMed
28.
Schuffler MD, Pagon RA, Schwartz R, et al. Visceral myopathy of the gastrointestinal and genitourinary tracts in infants. Gastroenterology. 1988;94:892–8.PubMed
29.
Martin JE, Benson M, Swash M, et al. Myofibroblasts in hollow visceral myopathy: the origin of gastrointestinal fibrosis? Gut. 1993;34:999–1001.PubMedCentralPubMed
30.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


